

Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes
- PMID: 23132910
- PMCID: PMC3561863
- DOI: 10.1101/gr.140301.112
Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes
Abstract
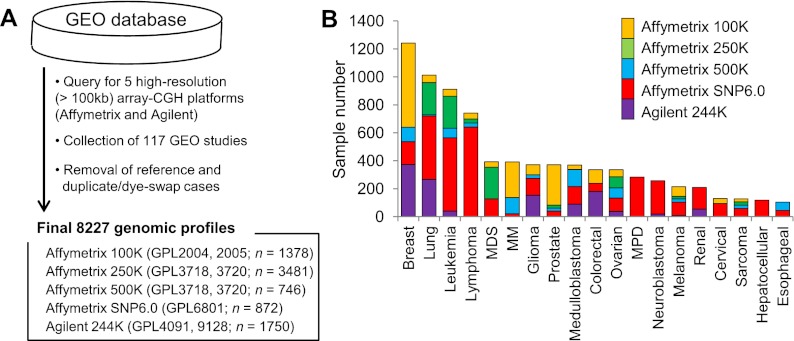
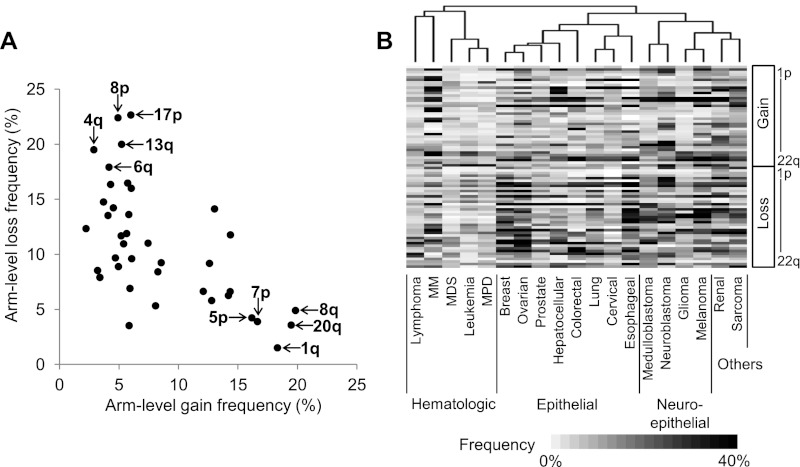
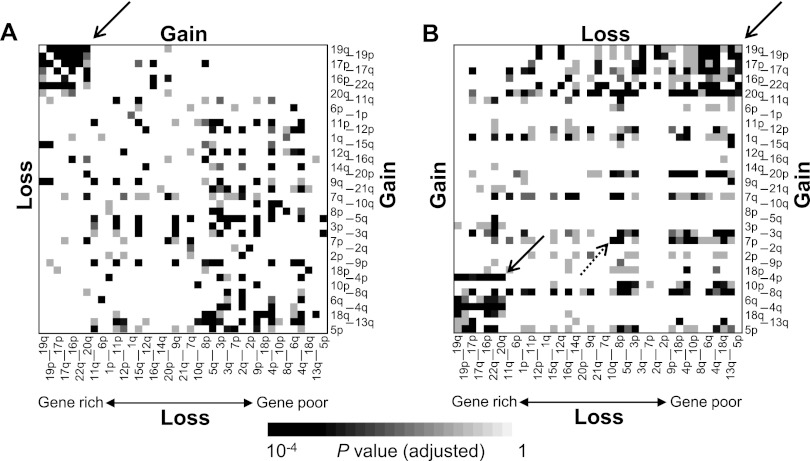
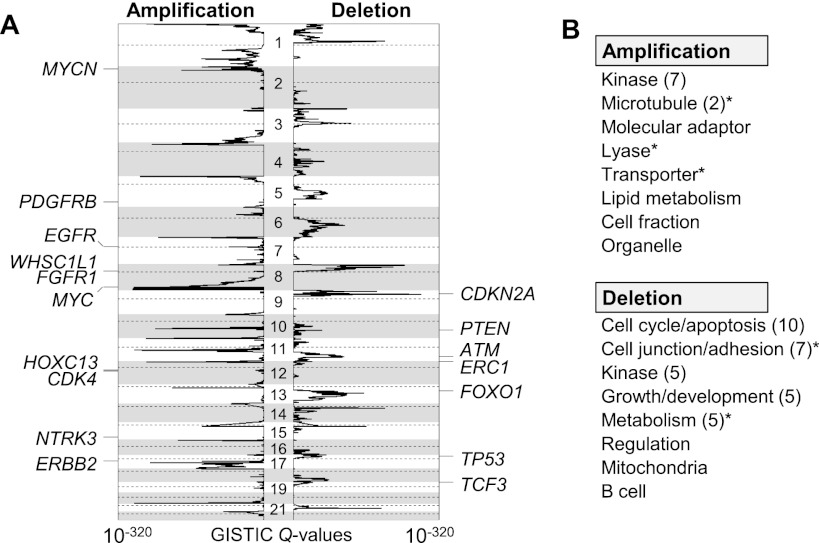
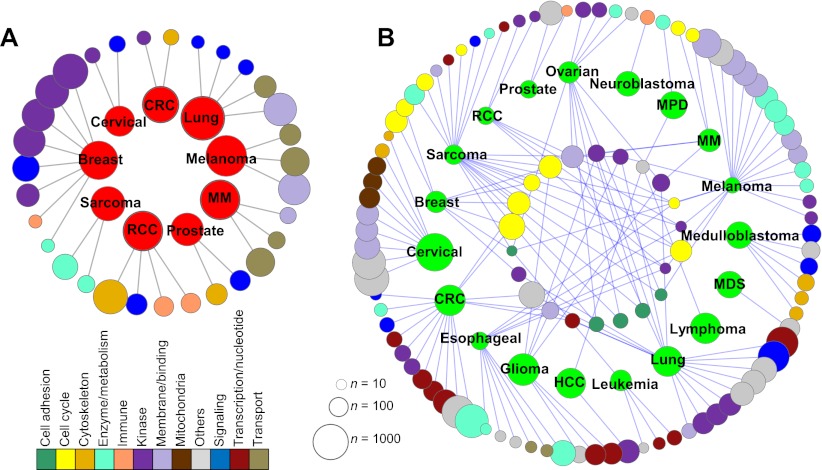
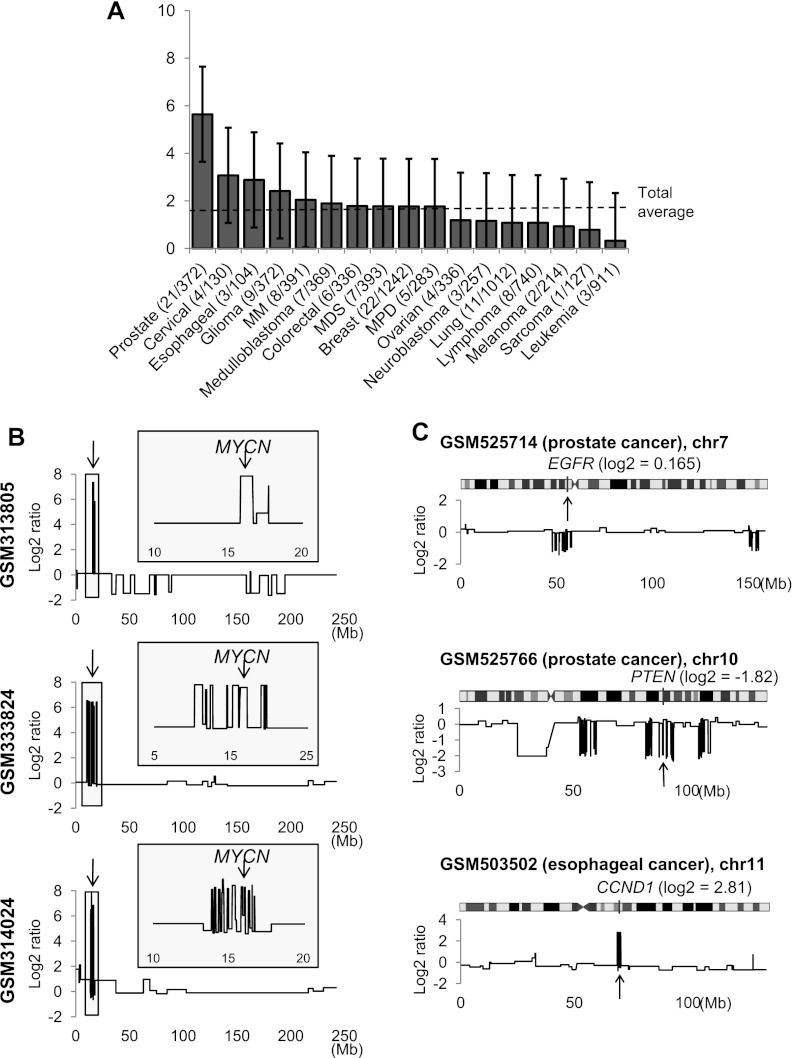
A large database of copy number profiles from cancer genomes can facilitate the identification of recurrent chromosomal alterations that often contain key cancer-related genes. It can also be used to explore low-prevalence genomic events such as chromothripsis. In this study, we report an analysis of 8227 human cancer copy number profiles obtained from 107 array comparative genomic hybridization (CGH) studies. Our analysis reveals similarity of chromosomal arm-level alterations among developmentally related tumor types as well as a number of co-occurring pairs of arm-level alterations. Recurrent ("pan-lineage") focal alterations identified across diverse tumor types show an enrichment of known cancer-related genes and genes with relevant functions in cancer-associated phenotypes (e.g., kinase and cell cycle). Tumor type-specific ("lineage-restricted") alterations and their enriched functional categories were also identified. Furthermore, we developed an algorithm for detecting regions in which the copy number oscillates rapidly between fixed levels, indicative of chromothripsis. We observed these massive genomic rearrangements in 1%-2% of the samples with variable tumor type-specific incidence rates. Taken together, our comprehensive view of copy number alterations provides a framework for understanding the functional significance of various genomic alterations in cancer genomes.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources