

Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures
- PMID: 23133383
- PMCID: PMC3486912
- DOI: 10.1371/journal.ppat.1002985
Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures
Abstract
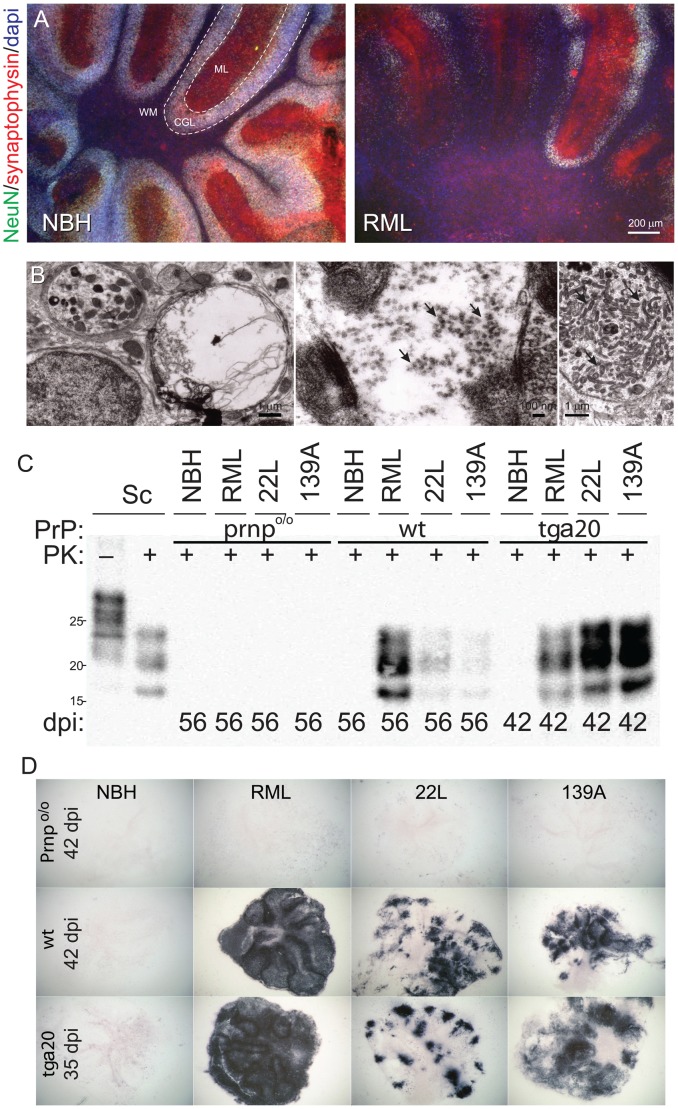
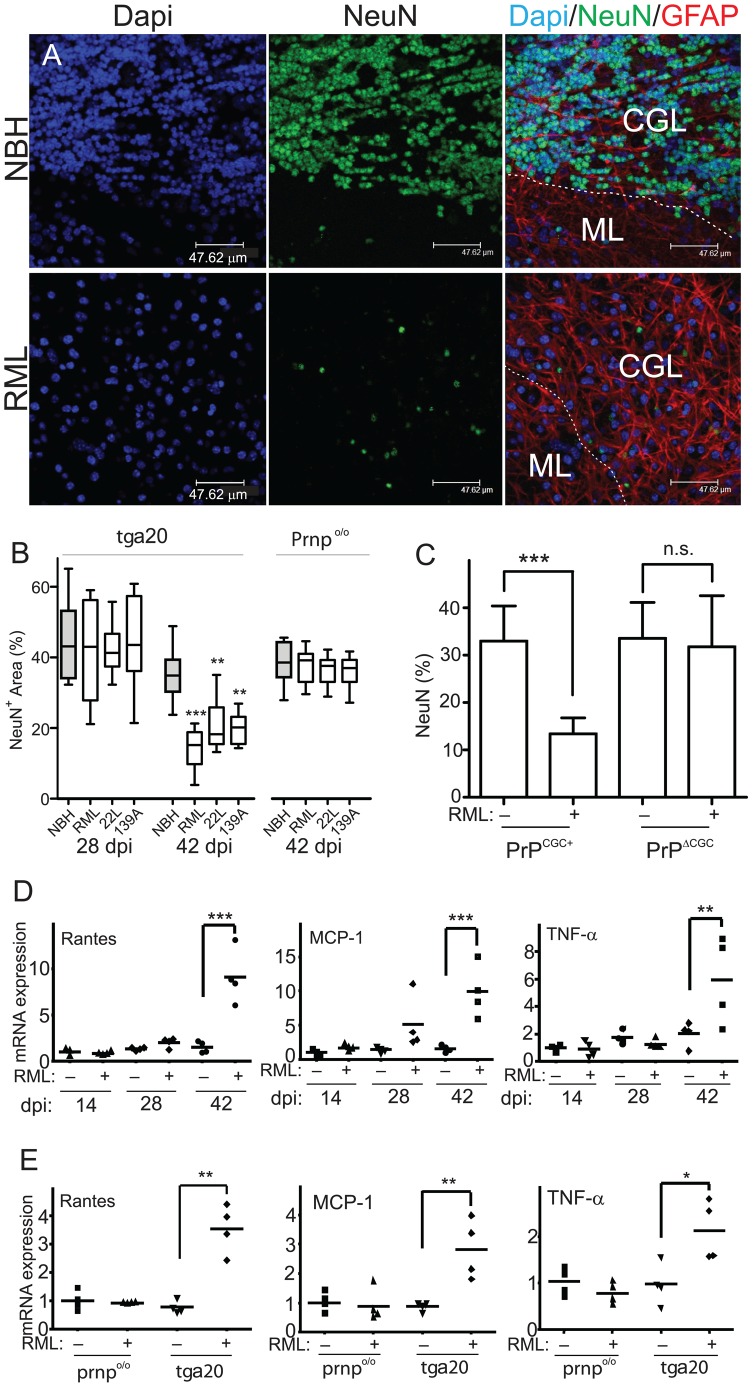
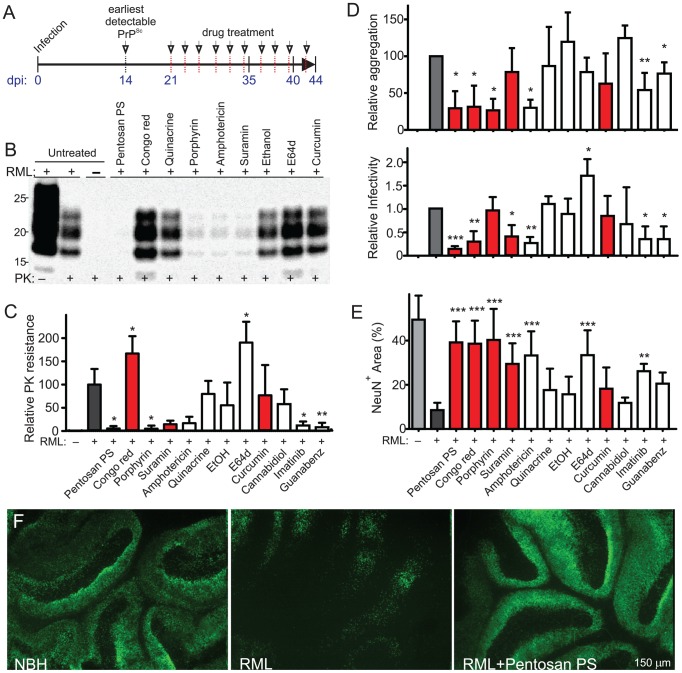
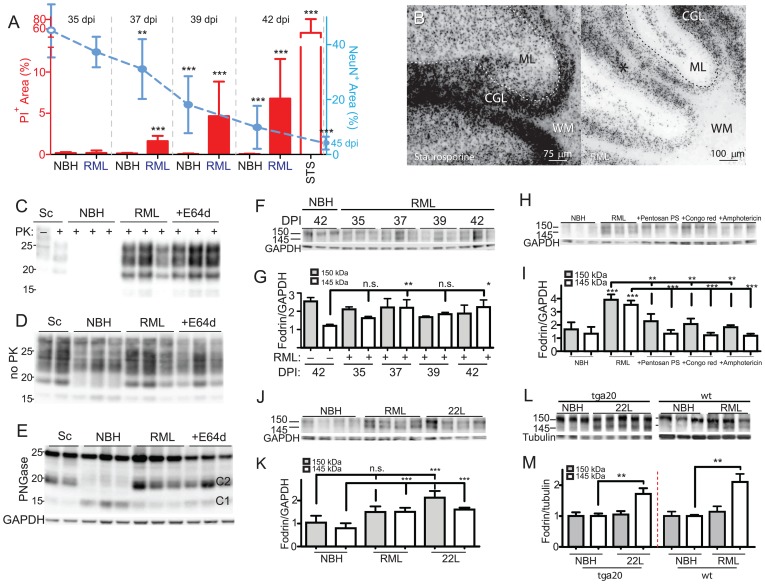
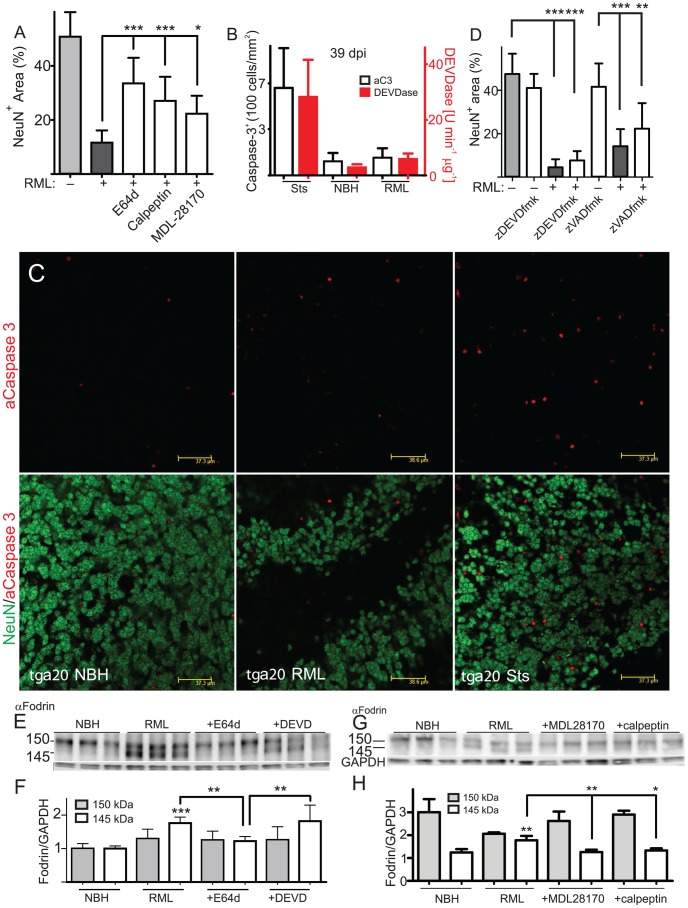
Prions cause neurodegeneration in vivo, yet prion-infected cultured cells do not show cytotoxicity. This has hampered mechanistic studies of prion-induced neurodegeneration. Here we report that prion-infected cultured organotypic cerebellar slices (COCS) experienced progressive spongiform neurodegeneration closely reproducing prion disease, with three different prion strains giving rise to three distinct patterns of prion protein deposition. Neurodegeneration did not occur when PrP was genetically removed from neurons, and a comprehensive pharmacological screen indicated that neurodegeneration was abrogated by compounds known to antagonize prion replication. Prion infection of COCS and mice led to enhanced fodrin cleavage, suggesting the involvement of calpains or caspases in pathogenesis. Accordingly, neurotoxicity and fodrin cleavage were prevented by calpain inhibitors but not by caspase inhibitors, whereas prion replication proceeded unimpeded. Hence calpain inhibition can uncouple prion replication from its neurotoxic sequelae. These data validate COCS as a powerful model system that faithfully reproduces most morphological hallmarks of prion infections. The exquisite accessibility of COCS to pharmacological manipulations was instrumental in recognizing the role of calpains in neurotoxicity, and significantly extends the collection of tools necessary for rigorously dissecting prion pathogenesis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
NG2 glia protect against prion neurotoxicity by inhibiting microglia-to-neuron prostaglandin E2 signaling.Nat Neurosci. 2024 Aug;27(8):1534-1544. doi: 10.1038/s41593-024-01663-x. Epub 2024 May 27. Nat Neurosci. 2024. PMID: 38802591 Free PMC article.
-
A neuroprotective role for microglia in prion diseases.J Exp Med. 2016 May 30;213(6):1047-59. doi: 10.1084/jem.20151000. Epub 2016 May 16. J Exp Med. 2016. PMID: 27185853 Free PMC article.
-
A versatile prion replication assay in organotypic brain slices.Nat Neurosci. 2008 Jan;11(1):109-17. doi: 10.1038/nn2028. Epub 2007 Dec 9. Nat Neurosci. 2008. PMID: 18066056 Free PMC article.
-
Mechanisms of prion-induced neurodegeneration.Expert Rev Mol Med. 2016 Apr 8;18:e5. doi: 10.1017/erm.2016.8. Expert Rev Mol Med. 2016. PMID: 27055367 Review.
-
Prion neurotoxicity: insights from prion protein mutants.Curr Issues Mol Biol. 2010;12(2):51-61. Epub 2009 Sep 18. Curr Issues Mol Biol. 2010. PMID: 19767650 Free PMC article. Review.
Cited by
-
Manipulating the Prion Protein Gene Sequence and Expression Levels with CRISPR/Cas9.PLoS One. 2016 Apr 29;11(4):e0154604. doi: 10.1371/journal.pone.0154604. eCollection 2016. PLoS One. 2016. PMID: 27128441 Free PMC article.
-
The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein.Nature. 2013 Sep 5;501(7465):102-6. doi: 10.1038/nature12402. Epub 2013 Jul 31. Nature. 2013. PMID: 23903654
-
Activation of pro-survival CaMK4β/CREB and pro-death MST1 signaling at early and late times during a mouse model of prion disease.Virol J. 2014 Sep 2;11:160. doi: 10.1186/1743-422X-11-160. Virol J. 2014. PMID: 25183307 Free PMC article.
-
From Cell Culture to Organoids-Model Systems for Investigating Prion Strain Characteristics.Biomolecules. 2021 Jan 14;11(1):106. doi: 10.3390/biom11010106. Biomolecules. 2021. PMID: 33466947 Free PMC article. Review.
-
Role of Prion Replication in the Strain-dependent Brain Regional Distribution of Prions.J Biol Chem. 2016 Jun 10;291(24):12880-12887. doi: 10.1074/jbc.M115.681791. Epub 2016 Apr 7. J Biol Chem. 2016. PMID: 27056328 Free PMC article.
References
-
- Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216: 136–144. - PubMed
-
- Büeler HR, Aguzzi A, Sailer A, Greiner RA, Autenried P, et al. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339–1347. - PubMed
-
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, et al. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326: 1533–1537. - PubMed
-
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, et al. (2010) Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13: 310–318. - PubMed
-
- Büeler HR, Fischer M, Lang Y, Bluethmann H, Lipp HP, et al. (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–582. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
