

Prader-willi syndrome: clinical aspects
- PMID: 23133744
- PMCID: PMC3486015
- DOI: 10.1155/2012/473941
Prader-willi syndrome: clinical aspects
Abstract
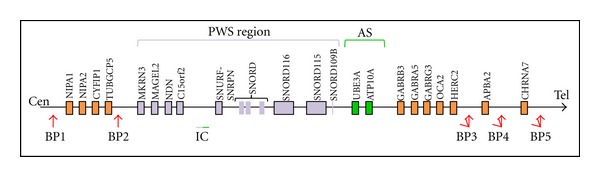
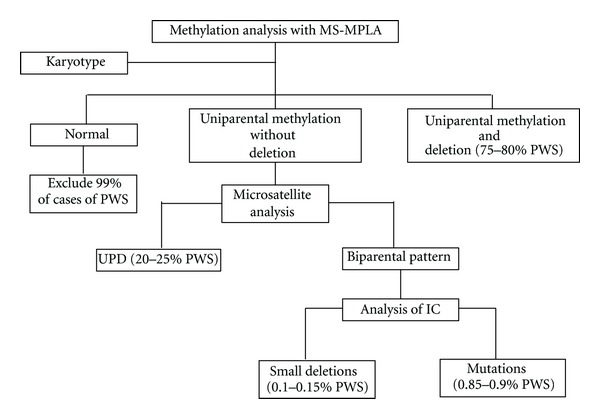
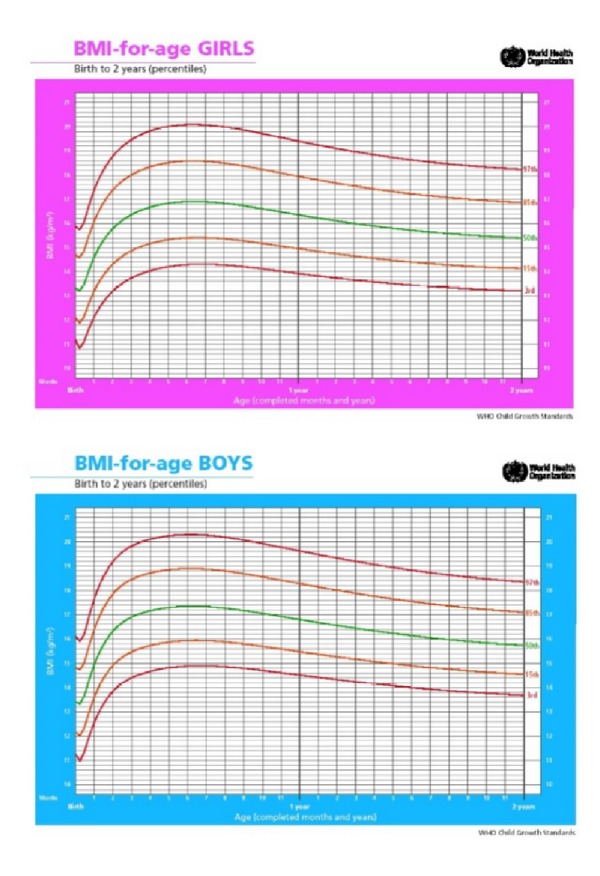
Prader-Willi Syndrome (PWS) is a complex multisystem genetic disorder that shows great variability, with changing clinical features during a patient's life. The syndrome is due to the loss of expression of several genes encoded on the proximal long arm of chromosome 15 (15q11.2-q13). The complex phenotype is most probably caused by a hypothalamic dysfunction that is responsible for hormonal dysfunctions and for absence of the sense of satiety. For this reason a Prader-Willi (PW) child develops hyperphagia during the initial stage of infancy that can lead to obesity and its complications. During infancy many PW child display a range of behavioural problems that become more noticeable in adolescence and adulthood and interfere mostly with quality of life. Early diagnosis of PWS is important for effective long-term management, and a precocious multidisciplinary approach is fundamental to improve quality of life, prevent complications, and prolong life expectancy.
Figures
Similar articles
-
Systematic review of the clinical and genetic aspects of Prader-Willi syndrome.Korean J Pediatr. 2011 Feb;54(2):55-63. doi: 10.3345/kjp.2011.54.2.55. Epub 2011 Feb 28. Korean J Pediatr. 2011. PMID: 21503198 Free PMC article.
-
The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria.Pediatrics. 2001 Nov;108(5):E92. doi: 10.1542/peds.108.5.e92. Pediatrics. 2001. PMID: 11694676
-
Prader- Willi syndrome: An uptodate on endocrine and metabolic complications.Rev Endocr Metab Disord. 2019 Jun;20(2):239-250. doi: 10.1007/s11154-019-09502-2. Rev Endocr Metab Disord. 2019. PMID: 31065942 Review.
-
Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings.J Endocrinol Invest. 2015 Dec;38(12):1249-63. doi: 10.1007/s40618-015-0312-9. Epub 2015 Jun 11. J Endocrinol Invest. 2015. PMID: 26062517 Free PMC article. Review.
-
Obesity management in Prader-Willi syndrome: current perspectives.Diabetes Metab Syndr Obes. 2018 Oct 4;11:579-593. doi: 10.2147/DMSO.S141352. eCollection 2018. Diabetes Metab Syndr Obes. 2018. PMID: 30323638 Free PMC article. Review.
Cited by
-
Hospital outcomes of scoliosis surgery in children with Prader-Willi Syndrome: comparison with adolescent idiopathic scoliosis.Spine Deform. 2021 Nov;9(6):1641-1647. doi: 10.1007/s43390-021-00359-7. Epub 2021 May 5. Spine Deform. 2021. PMID: 33950464
-
[Endocrine and metabolic features of female children with Prader-Willi syndrome: an analysis of 4 cases].Zhongguo Dang Dai Er Ke Za Zhi. 2017 May;19(5):514-518. doi: 10.7499/j.issn.1008-8830.2017.05.007. Zhongguo Dang Dai Er Ke Za Zhi. 2017. PMID: 28506340 Free PMC article. Chinese.
-
Oral disorders in children with Prader-Willi syndrome: a case control study.Orphanet J Rare Dis. 2020 Feb 10;15(1):43. doi: 10.1186/s13023-020-1326-8. Orphanet J Rare Dis. 2020. PMID: 32041633 Free PMC article.
-
Prader Willi syndrome and obstructive sleep apnea: co-occurrence in the pediatric population.J Clin Sleep Med. 2014 Apr 15;10(4):403-9. doi: 10.5664/jcsm.3616. J Clin Sleep Med. 2014. PMID: 24733986 Free PMC article.
-
Environmental Enrichment Normalizes Metabolic Function in the Murine Model of Prader-Willi Syndrome Magel2-Null Mice.Endocrinology. 2025 Feb 5;166(3):bqaf001. doi: 10.1210/endocr/bqaf001. Endocrinology. 2025. PMID: 39801003
References
-
- Prader A, Labhart A, Willi H. Ein syndrom von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myotonieartigem zustand im neugeborenenalter. Schweizerische Medizinische Wochenschrift. 1956;86:p. 1260.
-
- Driscoll DJ, Waters MF, Williams CA, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics. 1992;13(4):917–924. - PubMed
-
- Crinó A, Di Giorgio G, Livieri C, et al. A survey on Prader-Willi syndrome in the Italian population: prevalence of historical and clinical signs. Journal of Pediatric Endocrinology & Metabolism. 2009;22(10):883–893. - PubMed
-
- Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108(5, article E92) - PubMed
LinkOut - more resources
Full Text Sources
