

The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function
- PMID: 23136129
- PMCID: PMC3554197
- DOI: 10.1093/hmg/dds475
The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function
Abstract
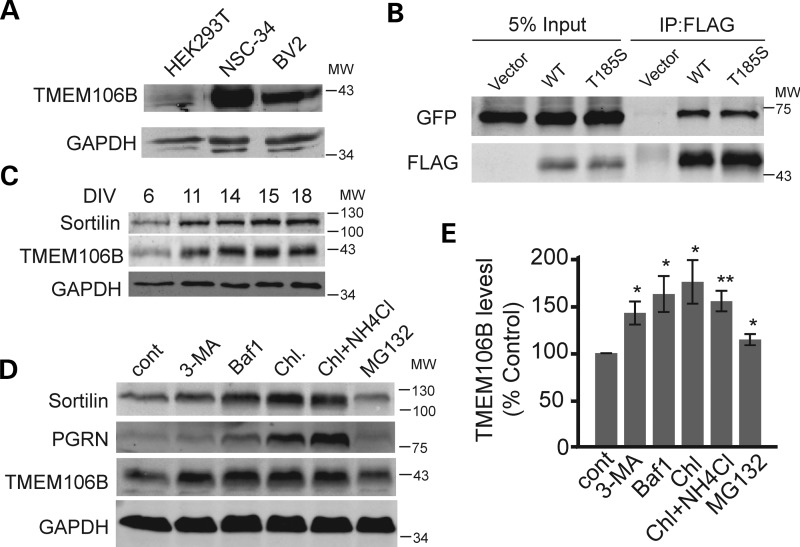
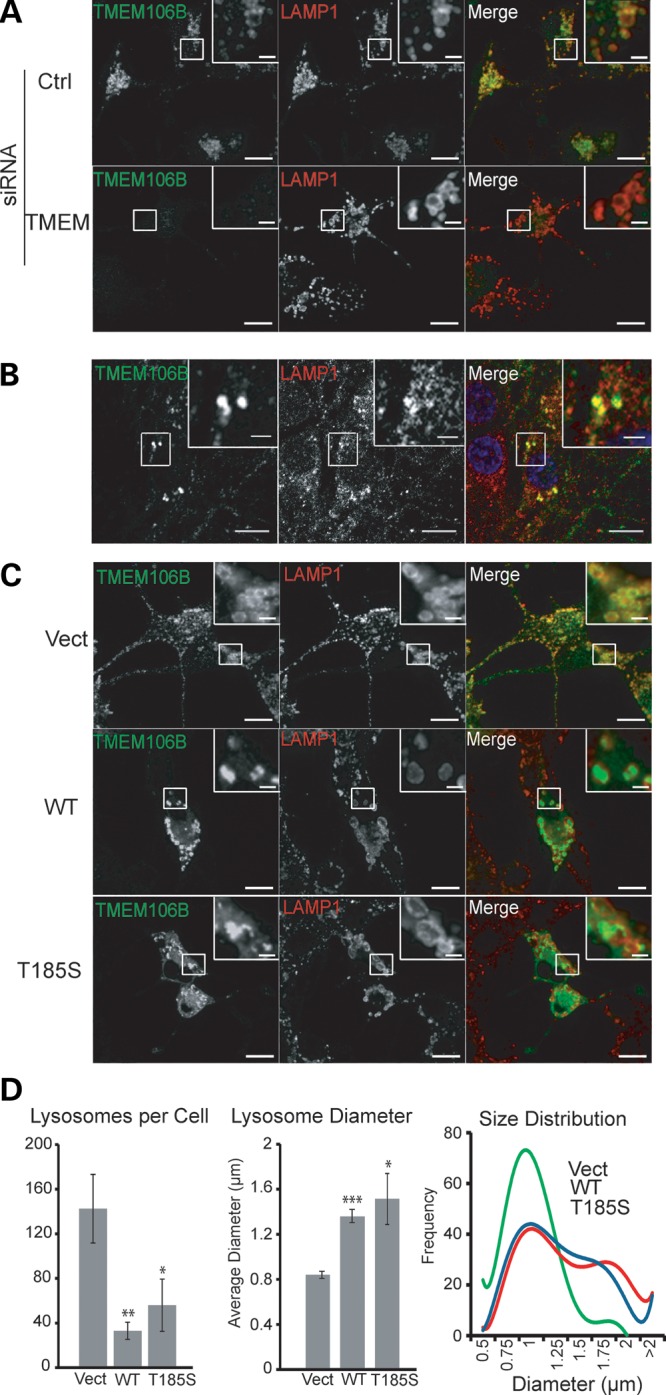
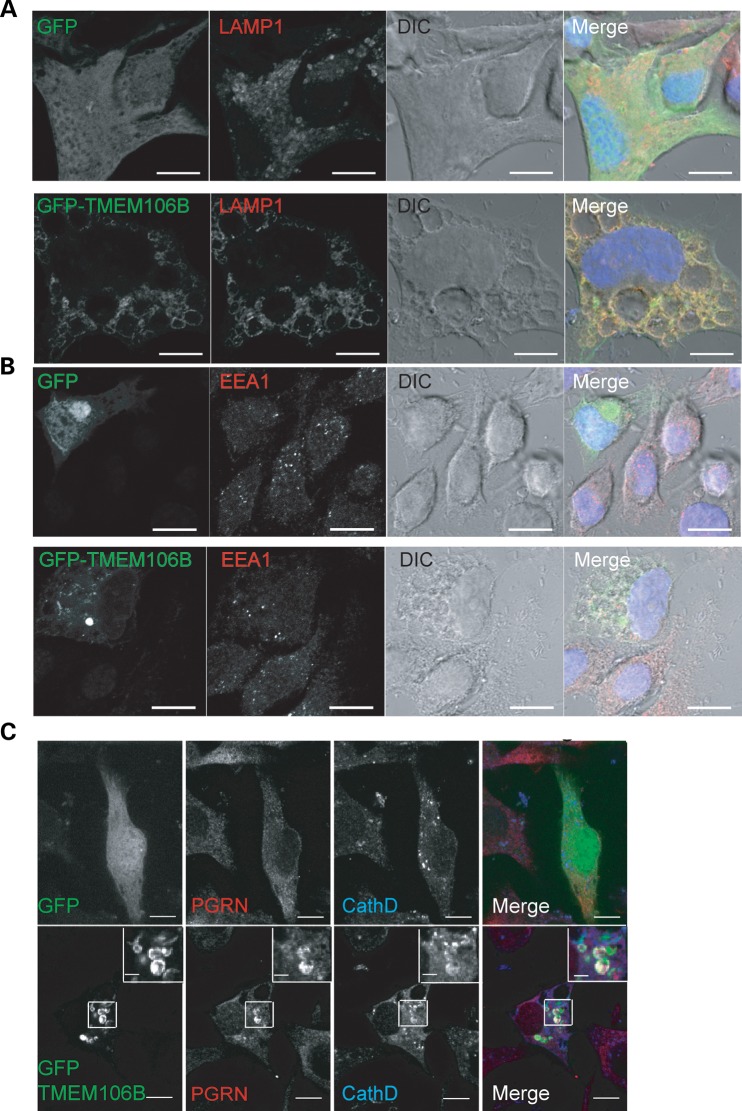
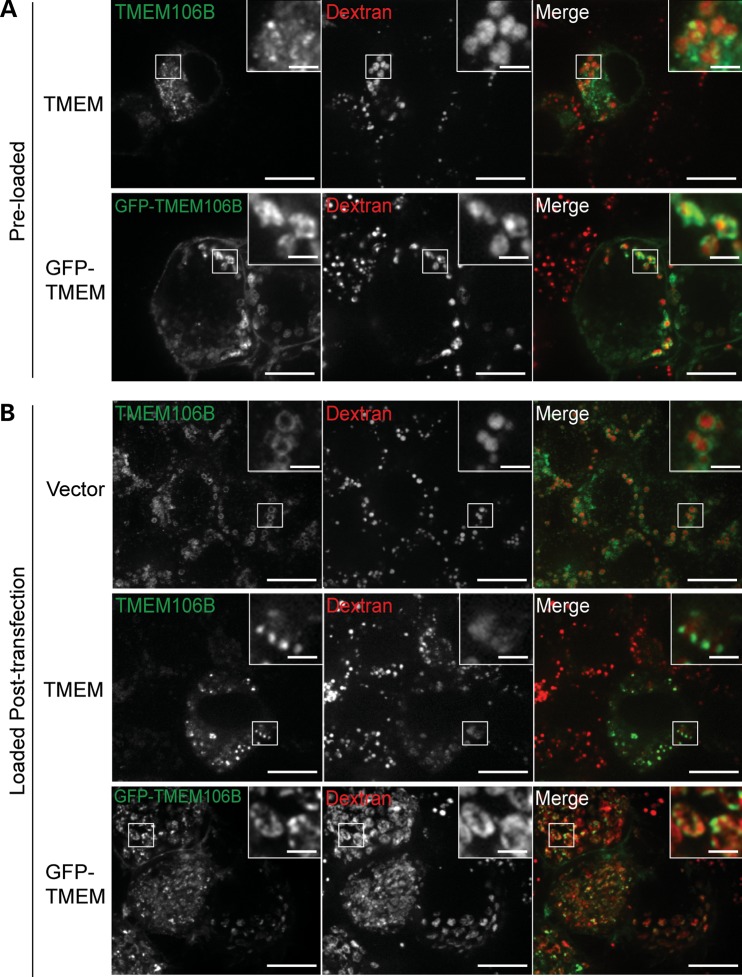
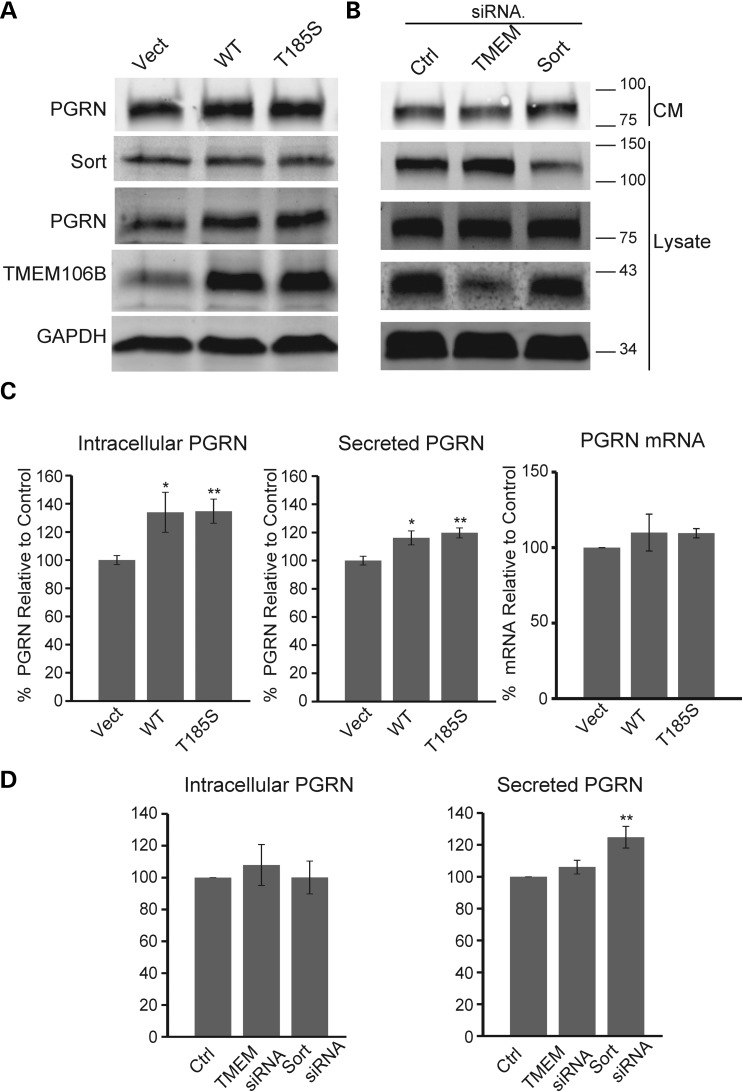
Haploinsufficiency of Progranulin (PGRN), a gene encoding a secreted glycoprotein, is a major cause of frontotemporal lobar degeneration with ubiquitin (FTLD-U) positive inclusions. Single nucleotide polymorphisms in the TMEM106B gene were recently discovered as a risk factor for FTLD-U, especially in patients with PGRN mutations. TMEM106B is also associated with cognitive impairment in amyotrophic lateral sclerosis patients. Despite these studies, little is known about TMEM106B at molecular and cellular levels and how TMEM106B contributes to FTLD. Here, we show that TMEM106B is localized in the late endosome/lysosome compartments and TMEM106B levels are regulated by lysosomal activities. Ectopic expression of TMEM106B induces morphologic changes of lysosome compartments and delays the degradation of endocytic cargoes by the endolysosomal pathway. Furthermore, overexpression of TMEM106B correlates with elevated levels of PGRN, possibly by attenuating lysosomal degradation of PGRN. These results shed light on the cellular functions of TMEM106B and the roles of TMEM106B in the pathogenesis of FTLD-U with PGRN mutations.
Figures

References
-
- Neary D., Snowden J., Mann D. Frontotemporal dementia. Lancet Neurol. 2005;4:771–780. - PubMed
-
- Ratnavalli E., Brayne C., Dawson K., Hodges J.R. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. - PubMed
-
- Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Cruts M., Gijselinck I., van der Zee J., Engelborghs S., Wils H., Pirici D., Rademakers R., Vandenberghe R., Dermaut B., Martin J.J., et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. - PubMed
-
- Gass J., Cannon A., Mackenzie I.R., Boeve B., Baker M., Adamson J., Crook R., Melquist S., Kuntz K., Petersen R., et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum. Mol. Genet. 2006;15:2988–3001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
