

Increased frequency of single base substitutions in a population of transcripts expressed in cancer cells
- PMID: 23137041
- PMCID: PMC3522053
- DOI: 10.1186/1471-2407-12-509
Increased frequency of single base substitutions in a population of transcripts expressed in cancer cells
Abstract
Background: Single Base Substitutions (SBS) that alter transcripts expressed in cancer originate from somatic mutations. However, recent studies report SBS in transcripts that are not supported by the genomic DNA of tumor cells.
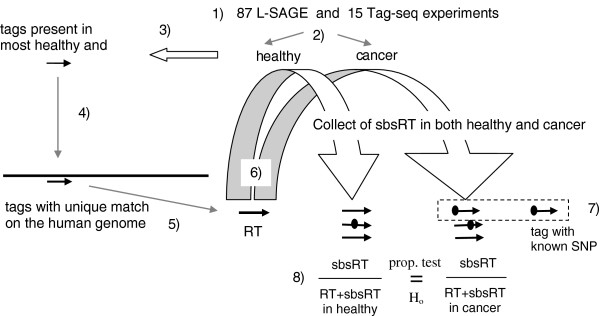
Methods: We used sequence based whole genome expression profiling, namely Long-SAGE (L-SAGE) and Tag-seq (a combination of L-SAGE and deep sequencing), and computational methods to identify transcripts with greater SBS frequencies in cancer. Millions of tags produced by 40 healthy and 47 cancer L-SAGE experiments were compared to 1,959 Reference Tags (RT), i.e. tags matching the human genome exactly once. Similarly, tens of millions of tags produced by 7 healthy and 8 cancer Tag-seq experiments were compared to 8,572 RT. For each transcript, SBS frequencies in healthy and cancer cells were statistically tested for equality.
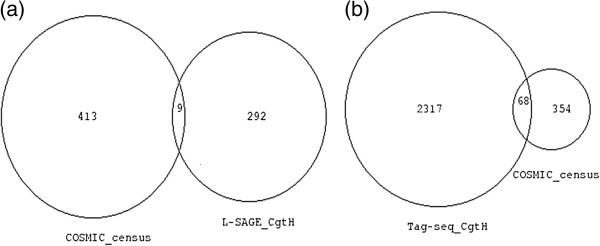
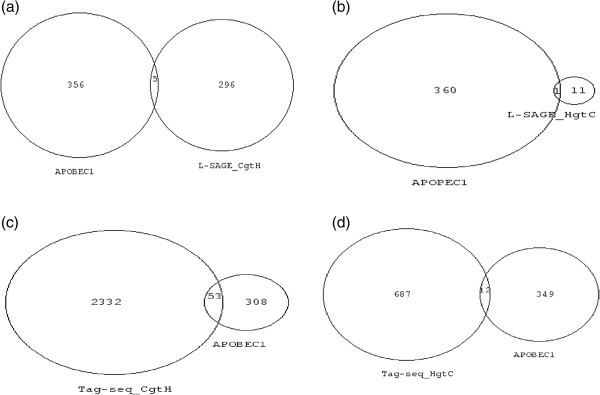
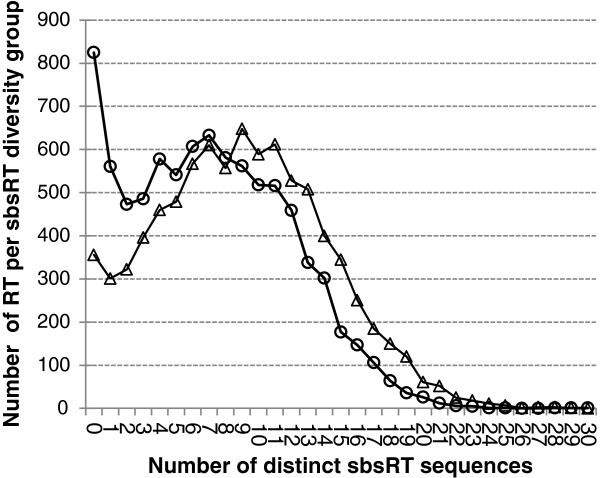
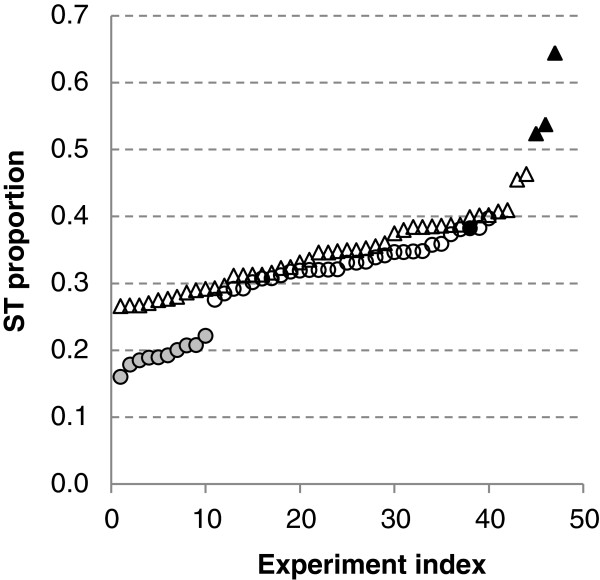
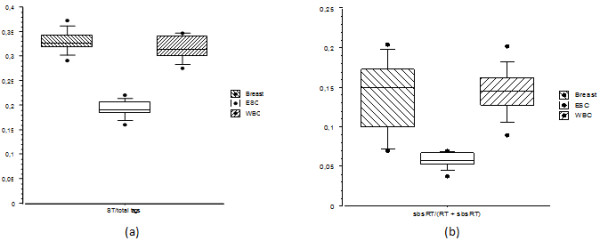
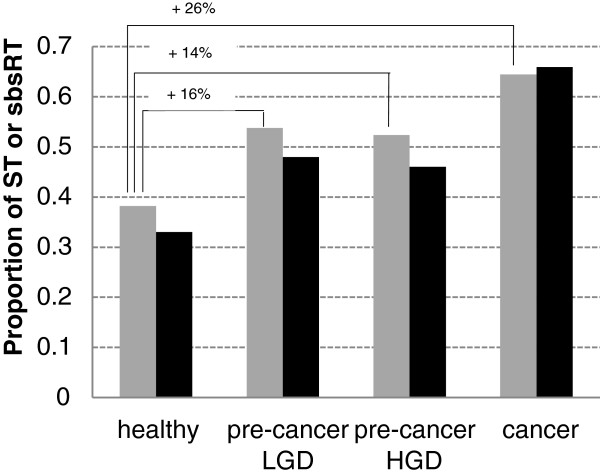
Results: In the L-SAGE and Tag-seq experiments, 372 and 4,289 transcripts respectively, showed greater SBS frequencies in cancer. Increased SBS frequencies could not be attributed to known Single Nucleotide Polymorphisms (SNP), catalogued somatic mutations or RNA-editing enzymes. Hypothesizing that Single Tags (ST), i.e. tags sequenced only once, were indicators of SBS, we observed that ST proportions were heterogeneously distributed across Embryonic Stem Cells (ESC), healthy differentiated and cancer cells. ESC had the lowest ST proportions, whereas cancer cells had the greatest. Finally, in a series of experiments carried out on a single patient at 1 healthy and 3 consecutive tumor stages, we could show that SBS frequencies increased during cancer progression.
Conclusion: If the mechanisms generating the base substitutions could be known, increased SBS frequency in transcripts would be a new useful biomarker of cancer. With the reduction of sequencing cost, sequence based whole genome expression profiling could be used to characterize increased SBS frequency in patient's tumor and aid diagnostic.
Figures
Similar articles
-
Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags.Proc Natl Acad Sci U S A. 2002 Sep 17;99(19):12257-62. doi: 10.1073/pnas.192436499. Epub 2002 Sep 4. Proc Natl Acad Sci U S A. 2002. PMID: 12213963 Free PMC article.
-
Deaminase-Driven Reverse Transcription Mutagenesis in Oncogenesis: Critical Analysis of Transcriptional Strand Asymmetries of Single Base Substitution Signatures.Int J Mol Sci. 2025 Jan 24;26(3):989. doi: 10.3390/ijms26030989. Int J Mol Sci. 2025. PMID: 39940758 Free PMC article. Review.
-
SAGETTARIUS: a program to reduce the number of tags mapped to multiple transcripts and to plan SAGE sequencing stages.Nucleic Acids Res. 2007;35(18):e122. doi: 10.1093/nar/gkm648. Epub 2007 Sep 20. Nucleic Acids Res. 2007. PMID: 17884916 Free PMC article.
-
High mutability of the tumor suppressor genes RASSF1 and RBSP3 (CTDSPL) in cancer.PLoS One. 2009 May 29;4(5):e5231. doi: 10.1371/journal.pone.0005231. PLoS One. 2009. PMID: 19478941 Free PMC article.
-
The emerging role of RNA and DNA editing in cancer.Biochim Biophys Acta. 2014 Apr;1845(2):308-16. doi: 10.1016/j.bbcan.2014.03.001. Epub 2014 Mar 7. Biochim Biophys Acta. 2014. PMID: 24607277 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
