

Protein structure prediction from sequence variation
- PMID: 23138306
- PMCID: PMC4319528
- DOI: 10.1038/nbt.2419
Protein structure prediction from sequence variation
Abstract
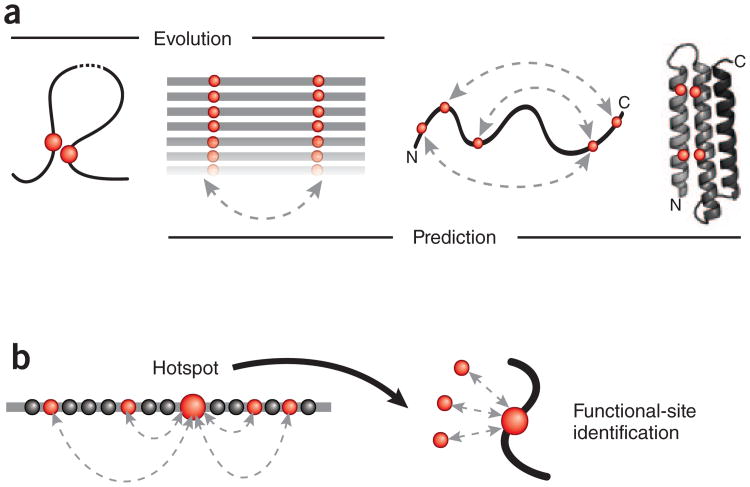
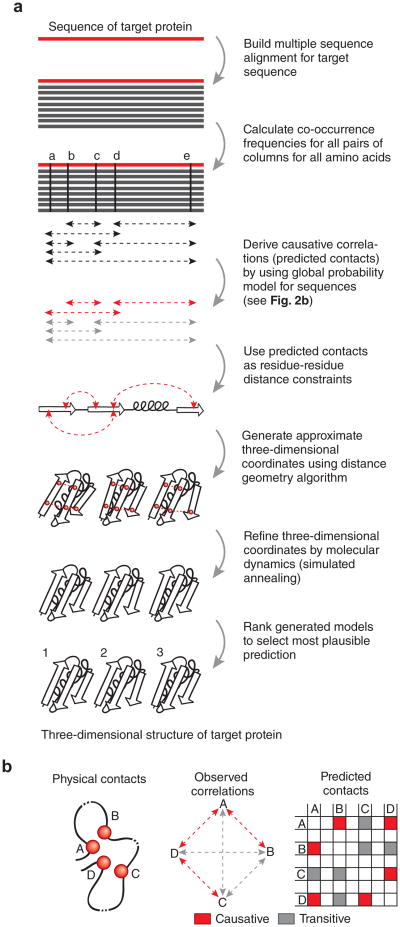
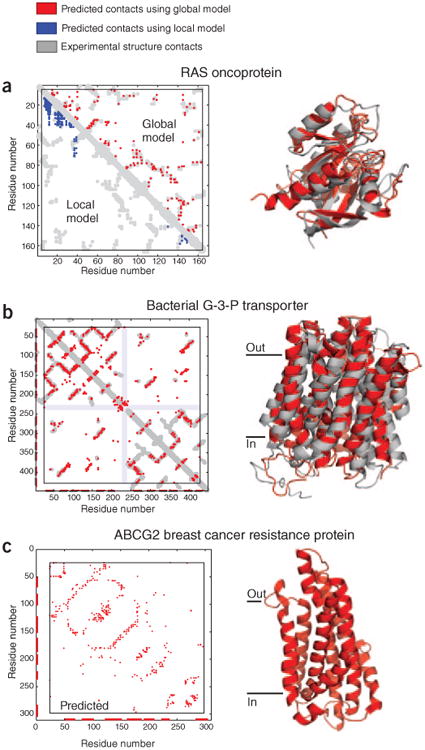
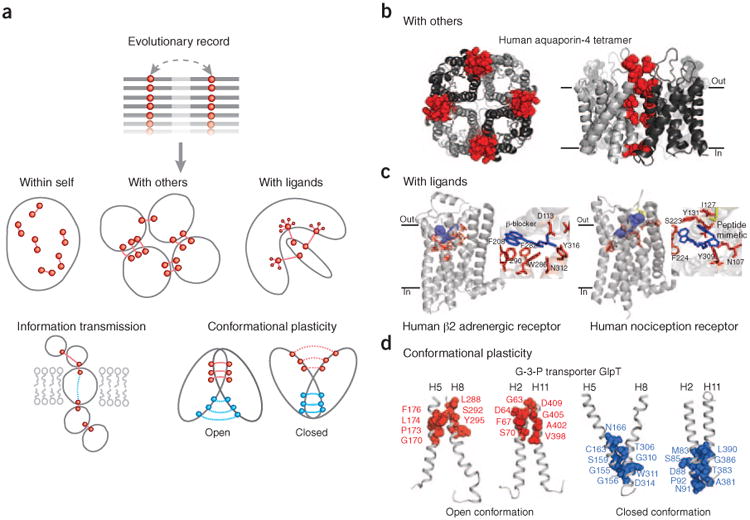
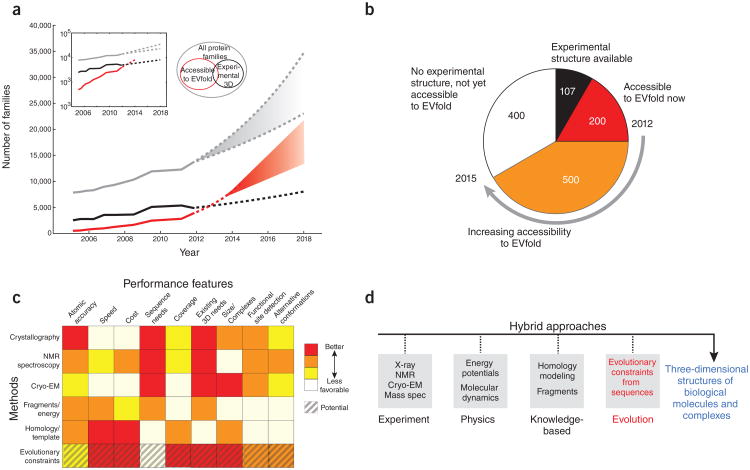
Genomic sequences contain rich evolutionary information about functional constraints on macromolecules such as proteins. This information can be efficiently mined to detect evolutionary couplings between residues in proteins and address the long-standing challenge to compute protein three-dimensional structures from amino acid sequences. Substantial progress has recently been made on this problem owing to the explosive growth in available sequences and the application of global statistical methods. In addition to three-dimensional structure, the improved understanding of covariation may help identify functional residues involved in ligand binding, protein-complex formation and conformational changes. We expect computation of covariation patterns to complement experimental structural biology in elucidating the full spectrum of protein structures, their functional interactions and evolutionary dynamics.
Figures
References
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. - PubMed
-
- Anfinsen CB. Some observations on the basic principles of design in protein molecules. Comp Biochem Physiol. 1962;4:229–240. - PubMed
-
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
