

Interpreting noncoding genetic variation in complex traits and human disease
- PMID: 23138309
- PMCID: PMC3703467
- DOI: 10.1038/nbt.2422
Interpreting noncoding genetic variation in complex traits and human disease
Abstract
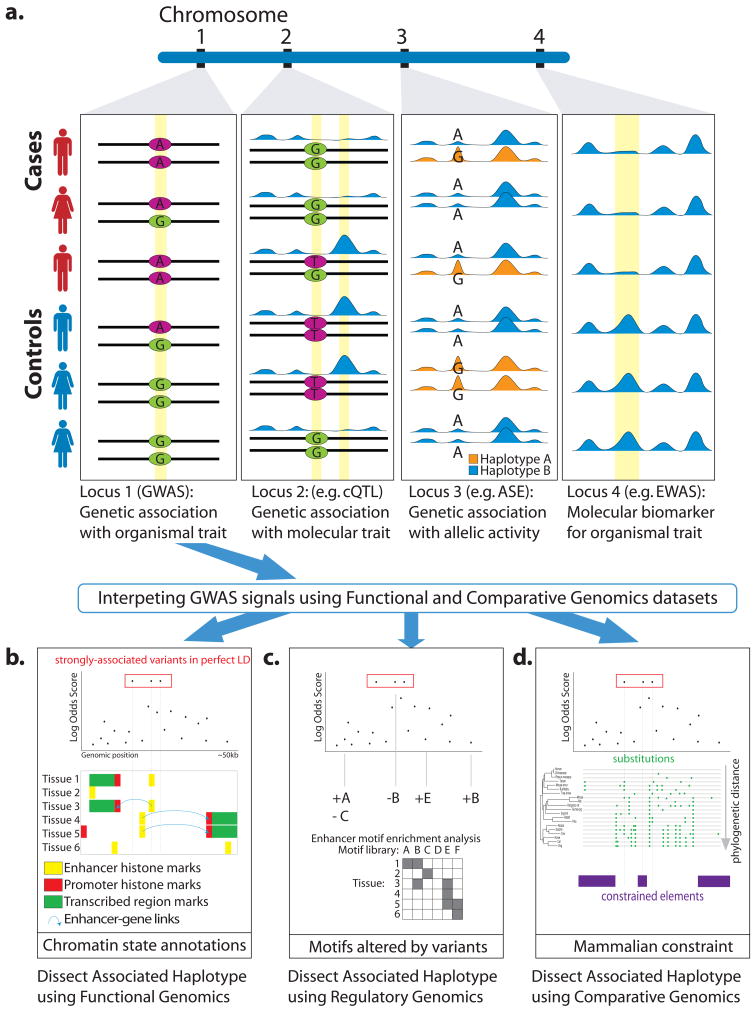
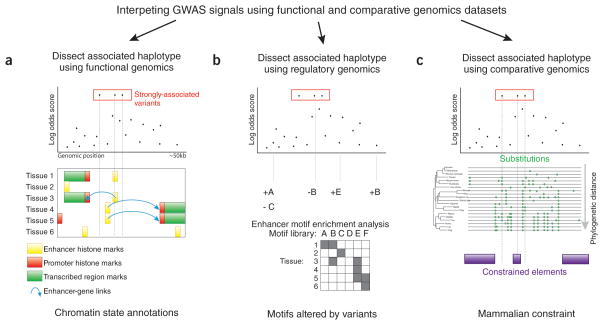
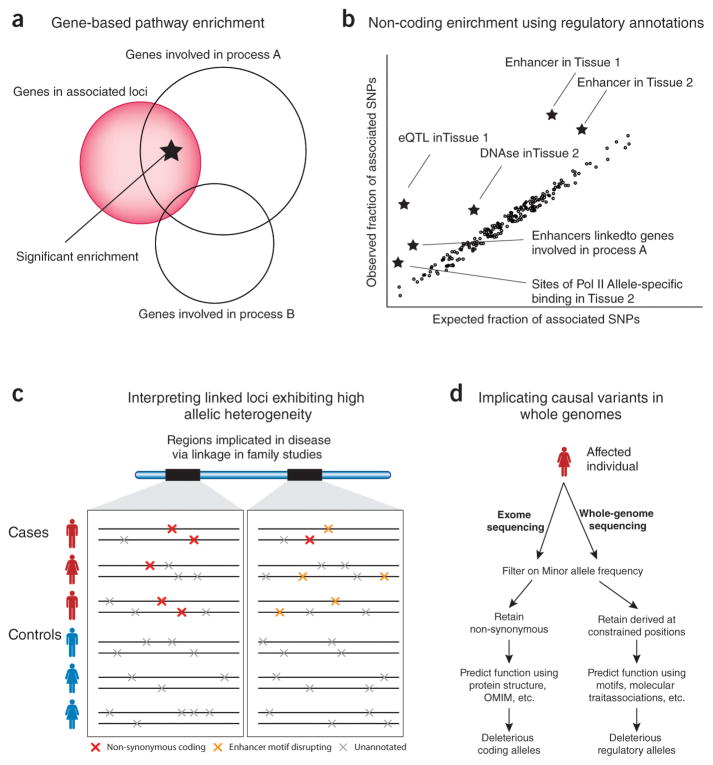
Association studies provide genome-wide information about the genetic basis of complex disease, but medical research has focused primarily on protein-coding variants, owing to the difficulty of interpreting noncoding mutations. This picture has changed with advances in the systematic annotation of functional noncoding elements. Evolutionary conservation, functional genomics, chromatin state, sequence motifs and molecular quantitative trait loci all provide complementary information about the function of noncoding sequences. These functional maps can help with prioritizing variants on risk haplotypes, filtering mutations encountered in the clinic and performing systems-level analyses to reveal processes underlying disease associations. Advances in predictive modeling can enable data-set integration to reveal pathways shared across loci and alleles, and richer regulatory models can guide the search for epistatic interactions. Lastly, new massively parallel reporter experiments can systematically validate regulatory predictions. Ultimately, advances in regulatory and systems genomics can help unleash the value of whole-genome sequencing for personalized genomic risk assessment, diagnosis and treatment.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
