

Current algorithmic solutions for peptide-based proteomics data generation and identification
- PMID: 23142544
- PMCID: PMC3857305
- DOI: 10.1016/j.copbio.2012.10.013
Current algorithmic solutions for peptide-based proteomics data generation and identification
Abstract
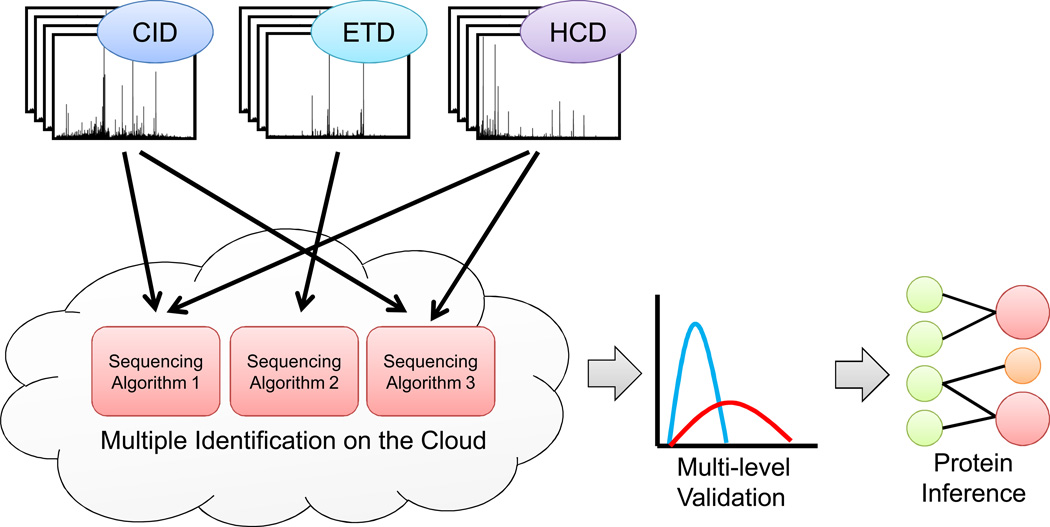
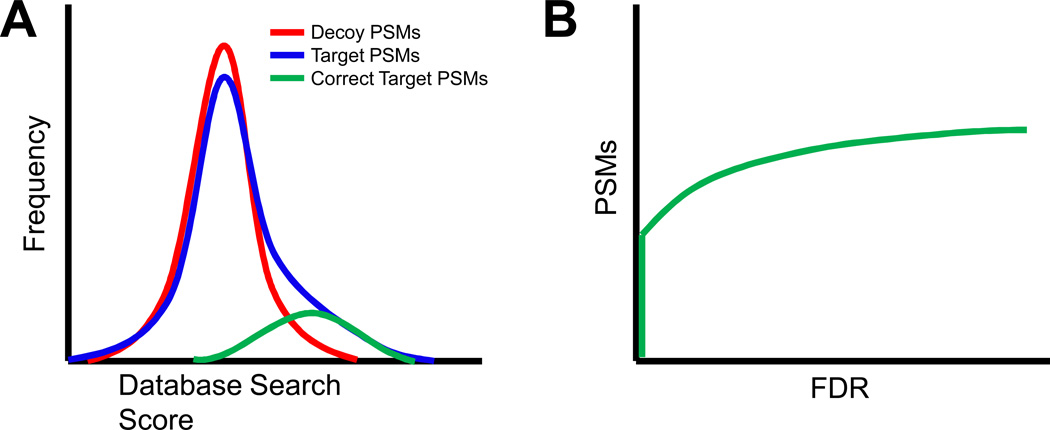
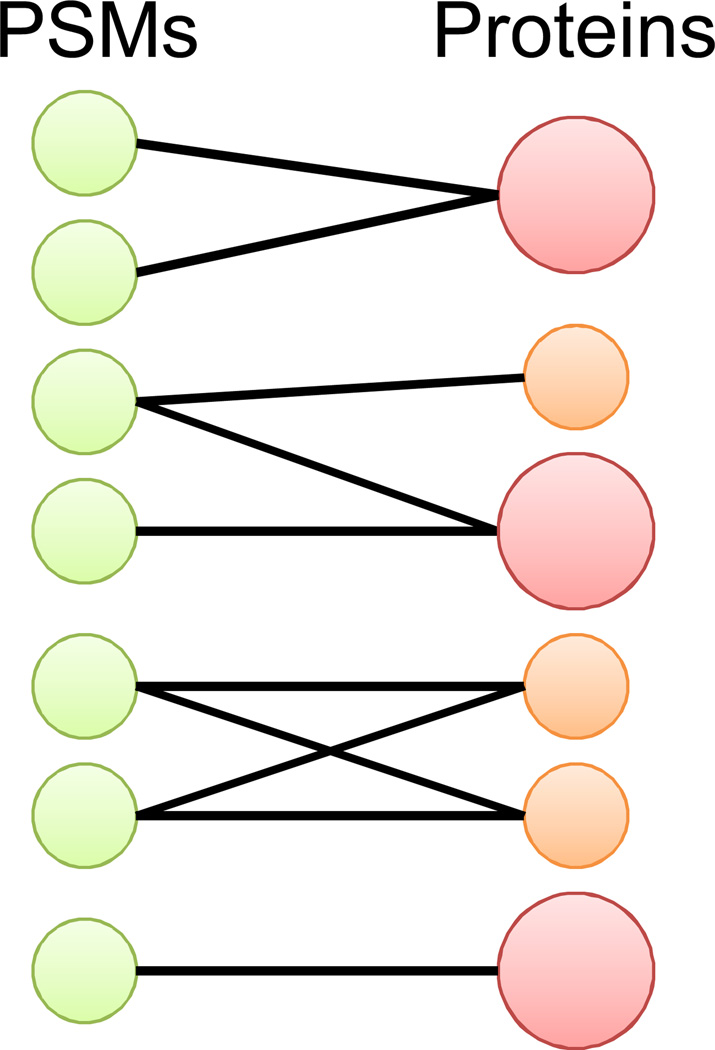
Peptide-based proteomic data sets are ever increasing in size and complexity. These data sets provide computational challenges when attempting to quickly analyze spectra and obtain correct protein identifications. Database search and de novo algorithms must consider high-resolution MS/MS spectra and alternative fragmentation methods. Protein inference is a tricky problem when analyzing large data sets of degenerate peptide identifications. Combining multiple algorithms for improved peptide identification puts significant strain on computational systems when investigating large data sets. This review highlights some of the recent developments in peptide and protein identification algorithms for analyzing shotgun mass spectrometry data when encountering the aforementioned hurdles. Also explored are the roles that analytical pipelines, public spectral libraries, and cloud computing play in the evolution of peptide-based proteomics.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
-
- Steen H, Mann M. The ABC's (and XYZ's) of peptide sequencing. Nat Rev Mol Cell Biol. 2004;5:699–711. - PubMed
-
- Frese CK, Altelaar AF, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJ, Mohammed S. Improved peptide identification by targeted fragmentation using CID HCD and ETD on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10:2377–2388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
