

Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity
- PMID: 23142757
- PMCID: PMC3529830
- DOI: 10.1016/j.chembiol.2012.09.019
Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity
Abstract
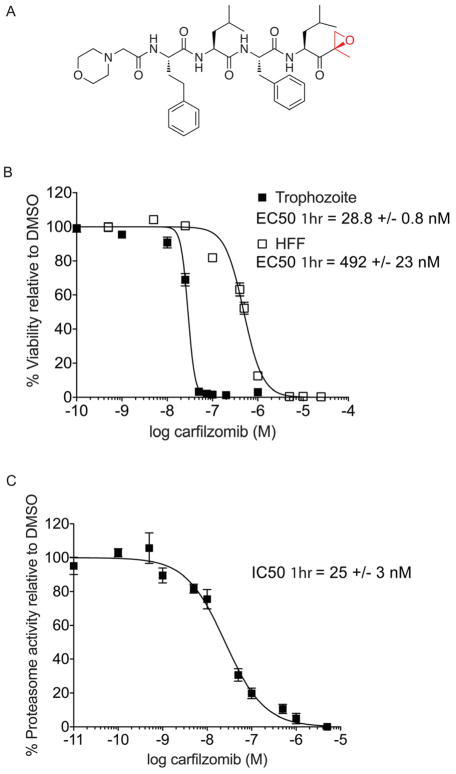
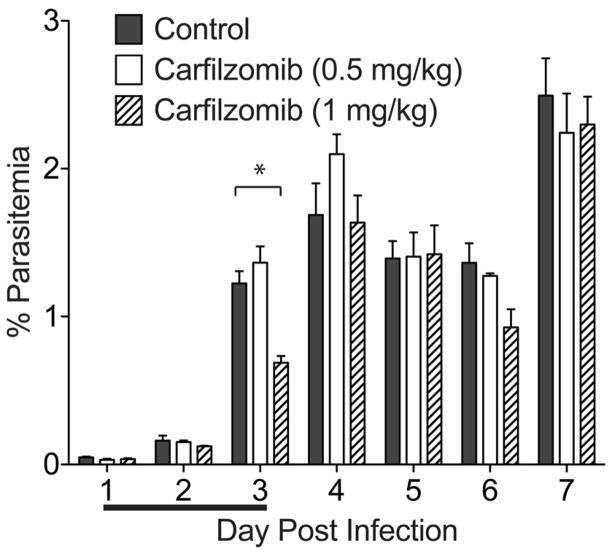
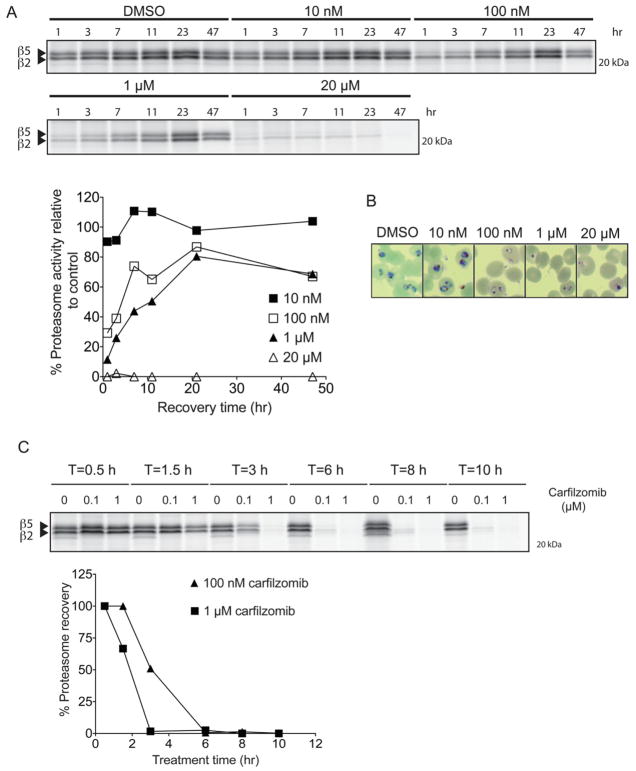
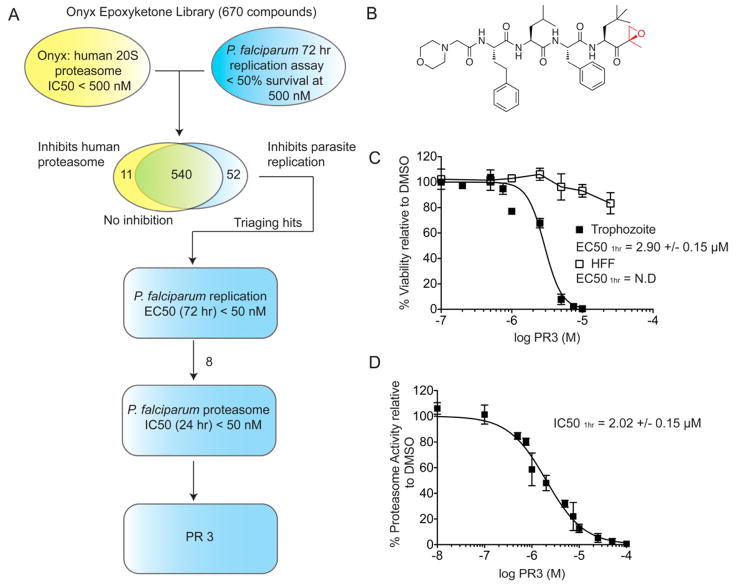
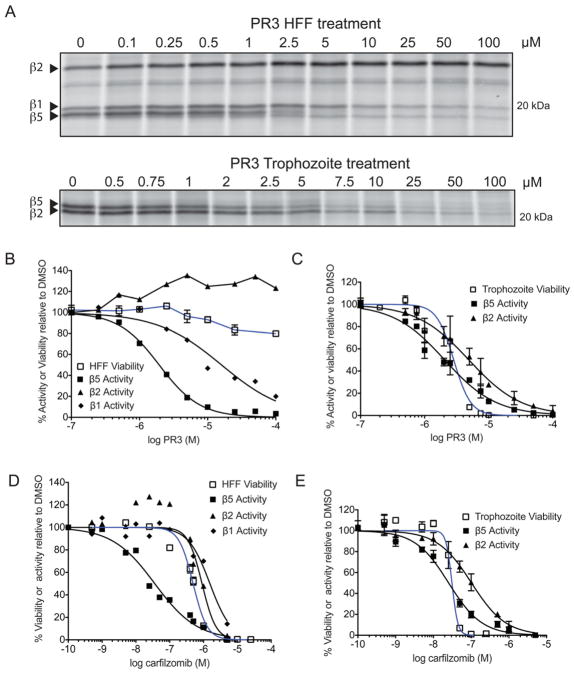
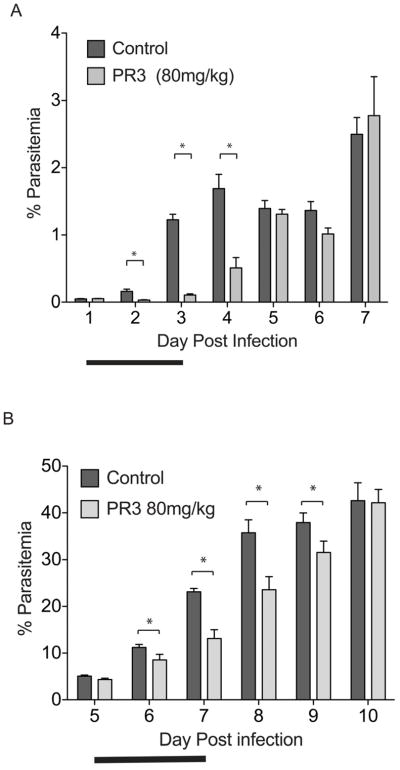
The Plasmodium proteasome has been suggested to be a potential antimalarial drug target; however, toxicity of inhibitors has prevented validation of this enzyme in vivo. We report a screen of a library of 670 analogs of the recent US Food and Drug Administration-approved inhibitor, carfilzomib, to identify compounds that selectively kill parasites. We identified one compound, PR3, that has significant parasite killing activity in vitro but dramatically reduced toxicity in host cells. We found that this parasite-specific toxicity is not due to selective targeting of the Plasmodium proteasome over the host proteasome, but instead is due to a lack of activity against one of the human proteasome subunits. Subsequently, we used PR3 to significantly reduce parasite load in Plasmodium berghei infected mice without host toxicity, thus validating the proteasome as a viable antimalarial drug target.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome.J Am Chem Soc. 2014 Oct 1;136(39):13562-5. doi: 10.1021/ja507692y. Epub 2014 Sep 19. J Am Chem Soc. 2014. PMID: 25226494 Free PMC article.
-
Structure- and function-based design of Plasmodium-selective proteasome inhibitors.Nature. 2016 Feb 11;530(7589):233-6. doi: 10.1038/nature16936. Nature. 2016. PMID: 26863983 Free PMC article.
-
Ubiquitin Proteasome System as a Potential Drug Target for Malaria.Curr Top Med Chem. 2018;18(5):315-320. doi: 10.2174/1568026618666180427145308. Curr Top Med Chem. 2018. PMID: 29701143 Review.
-
Mitigating the risk of antimalarial resistance via covalent dual-subunit inhibition of the Plasmodium proteasome.Cell Chem Biol. 2023 May 18;30(5):470-485.e6. doi: 10.1016/j.chembiol.2023.03.002. Epub 2023 Mar 23. Cell Chem Biol. 2023. PMID: 36963402 Free PMC article.
-
The proteasome as a target to combat malaria: hits and misses.Transl Res. 2018 Aug;198:40-47. doi: 10.1016/j.trsl.2018.04.007. Epub 2018 May 3. Transl Res. 2018. PMID: 30009761 Free PMC article. Review.
Cited by
-
Profound activity of the anti-cancer drug bortezomib against Echinococcus multilocularis metacestodes identifies the proteasome as a novel drug target for cestodes.PLoS Negl Trop Dis. 2014 Dec 4;8(12):e3352. doi: 10.1371/journal.pntd.0003352. eCollection 2014 Dec. PLoS Negl Trop Dis. 2014. PMID: 25474446 Free PMC article.
-
Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome.J Am Chem Soc. 2014 Oct 1;136(39):13562-5. doi: 10.1021/ja507692y. Epub 2014 Sep 19. J Am Chem Soc. 2014. PMID: 25226494 Free PMC article.
-
Design of proteasome inhibitors with oral efficacy in vivo against Plasmodium falciparum and selectivity over the human proteasome.Proc Natl Acad Sci U S A. 2021 Sep 28;118(39):e2107213118. doi: 10.1073/pnas.2107213118. Proc Natl Acad Sci U S A. 2021. PMID: 34548400 Free PMC article.
-
Selective targeting of Plasmodium falciparum Hsp90 disrupts the 26S proteasome.Cell Chem Biol. 2024 Apr 18;31(4):729-742.e13. doi: 10.1016/j.chembiol.2024.02.008. Epub 2024 Mar 15. Cell Chem Biol. 2024. PMID: 38492573 Free PMC article.
-
Identification of potent and reversible piperidine carboxamides that are species-selective orally active proteasome inhibitors to treat malaria.Cell Chem Biol. 2024 Aug 15;31(8):1503-1517.e19. doi: 10.1016/j.chembiol.2024.07.001. Epub 2024 Jul 30. Cell Chem Biol. 2024. PMID: 39084225 Free PMC article.
References
-
- Arastu-Kapur S, Ponder EL, Fonović UP, Yeoh S, Yuan F, Fonović M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4:203–213. - PubMed
-
- Blackman MJ. Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Methods Cell Biol. 1994;45:213–220. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
