

ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth
- PMID: 23143306
- PMCID: PMC3533536
- DOI: 10.1172/JCI62973
ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth
Abstract
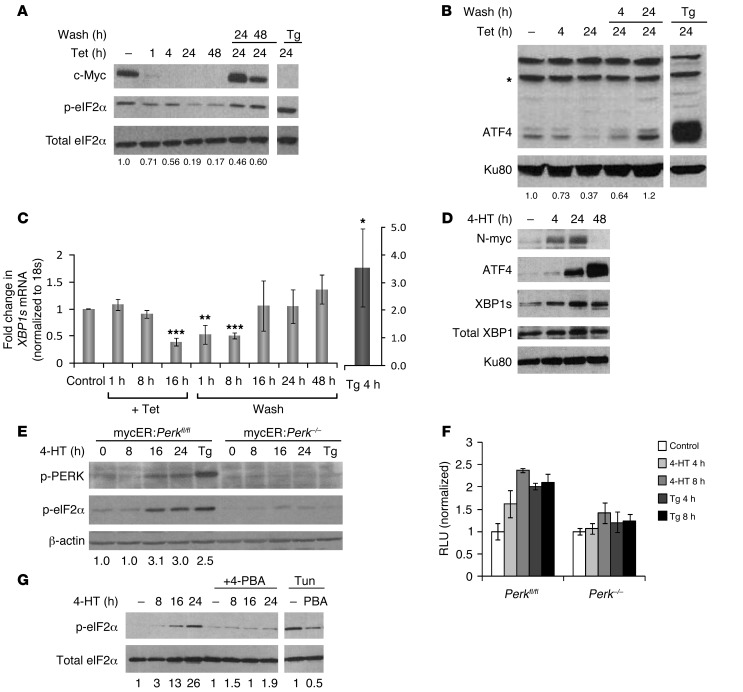
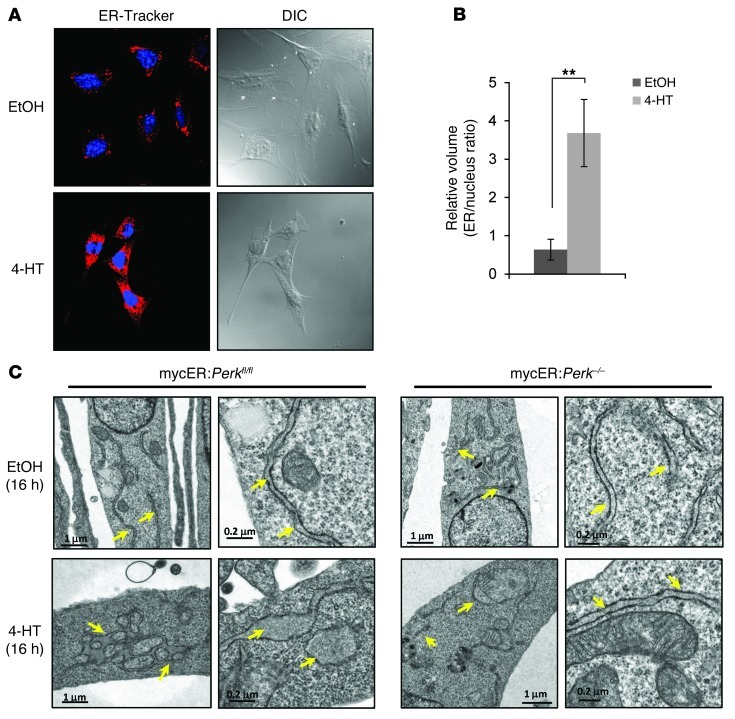
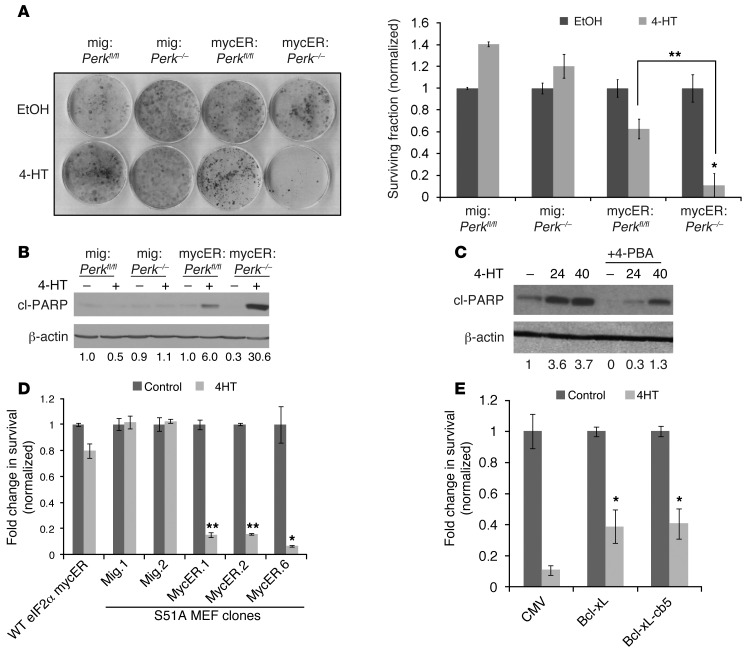
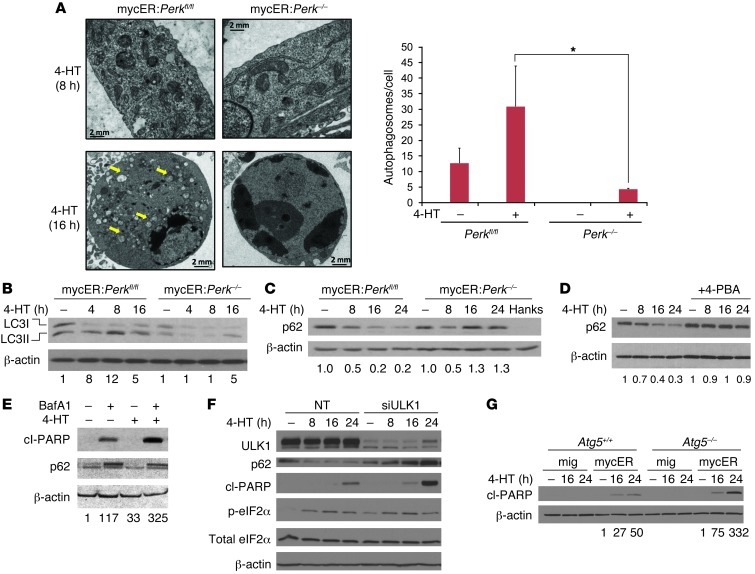
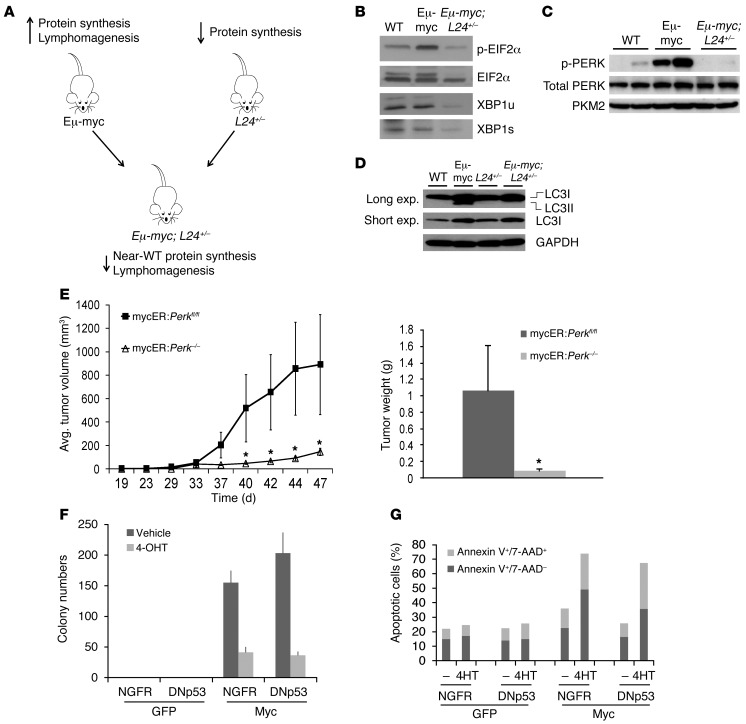
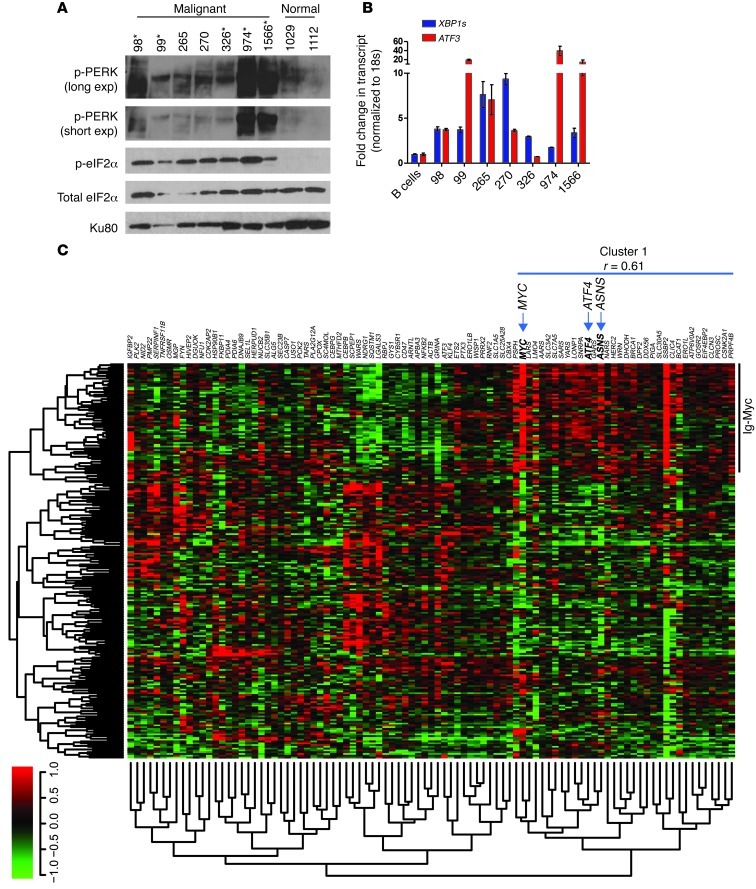
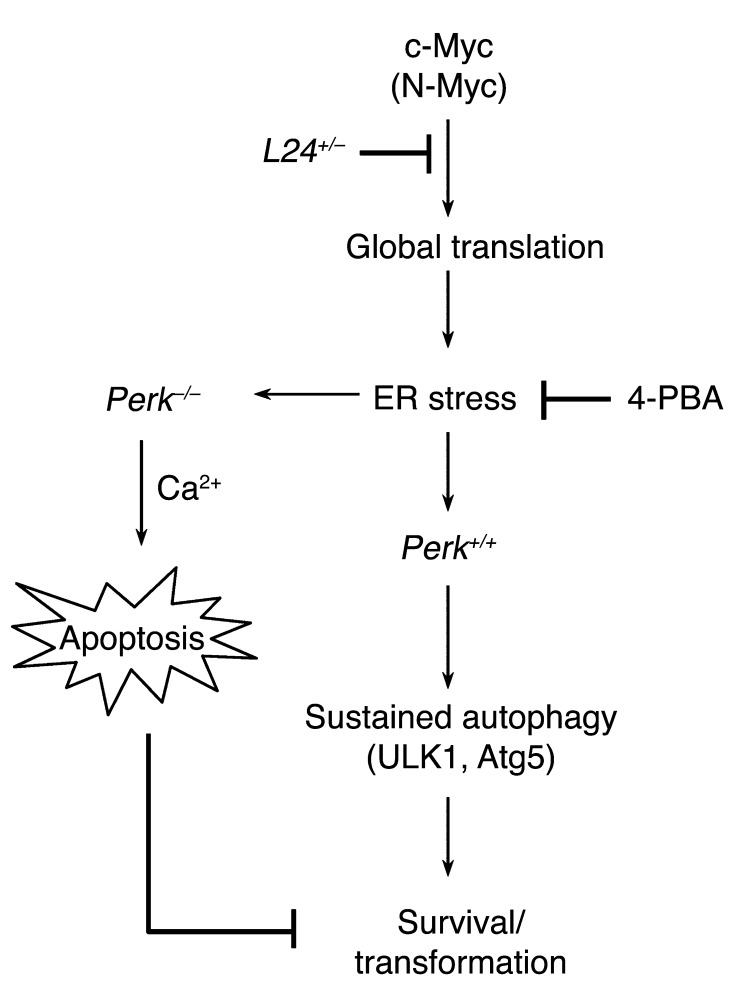
The proto-oncogene c-Myc paradoxically activates both proliferation and apoptosis. In the pathogenic state, c-Myc-induced apoptosis is bypassed via a critical, yet poorly understood escape mechanism that promotes cellular transformation and tumorigenesis. The accumulation of unfolded proteins in the ER initiates a cellular stress program termed the unfolded protein response (UPR) to support cell survival. Analysis of spontaneous mouse and human lymphomas demonstrated significantly higher levels of UPR activation compared with normal tissues. Using multiple genetic models, we demonstrated that c-Myc and N-Myc activated the PERK/eIF2α/ATF4 arm of the UPR, leading to increased cell survival via the induction of cytoprotective autophagy. Inhibition of PERK significantly reduced Myc-induced autophagy, colony formation, and tumor formation. Moreover, pharmacologic or genetic inhibition of autophagy resulted in increased Myc-dependent apoptosis. Mechanistically, we demonstrated an important link between Myc-dependent increases in protein synthesis and UPR activation. Specifically, by employing a mouse minute (L24+/-) mutant, which resulted in wild-type levels of protein synthesis and attenuation of Myc-induced lymphomagenesis, we showed that Myc-induced UPR activation was reversed. Our findings establish a role for UPR as an enhancer of c-Myc-induced transformation and suggest that UPR inhibition may be particularly effective against malignancies characterized by c-Myc overexpression.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- P01 CA165997/CA/NCI NIH HHS/United States
- R01CA122334/CA/NCI NIH HHS/United States
- R01 CA122334/CA/NCI NIH HHS/United States
- R01 CA092900/CA/NCI NIH HHS/United States
- R01CA094214/CA/NCI NIH HHS/United States
- P01 CA104838/CA/NCI NIH HHS/United States
- R01 CA094214/CA/NCI NIH HHS/United States
- R01CA140456/CA/NCI NIH HHS/United States
- R01 CA140456/CA/NCI NIH HHS/United States
- R01 CA154916/CA/NCI NIH HHS/United States
- R01 CA142425/CA/NCI NIH HHS/United States
- R01CA139362/CA/NCI NIH HHS/United States
- P01CA104838/CA/NCI NIH HHS/United States
- R01 CA139362/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
