

Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2
- PMID: 23143600
- PMCID: PMC3671095
- DOI: 10.1038/ng.2454
Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2
Abstract
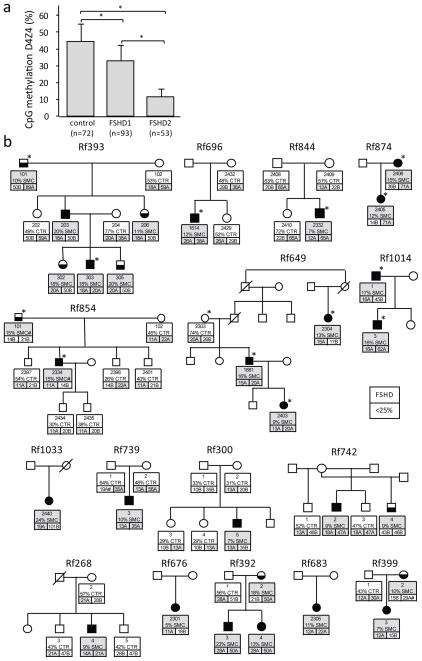
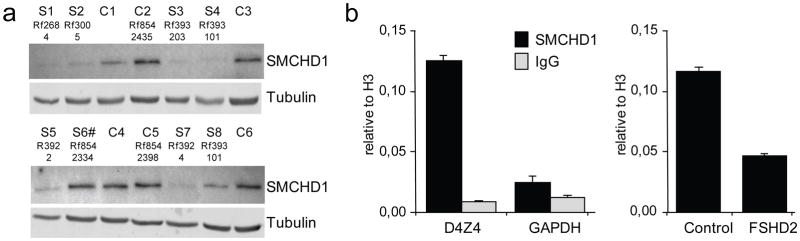
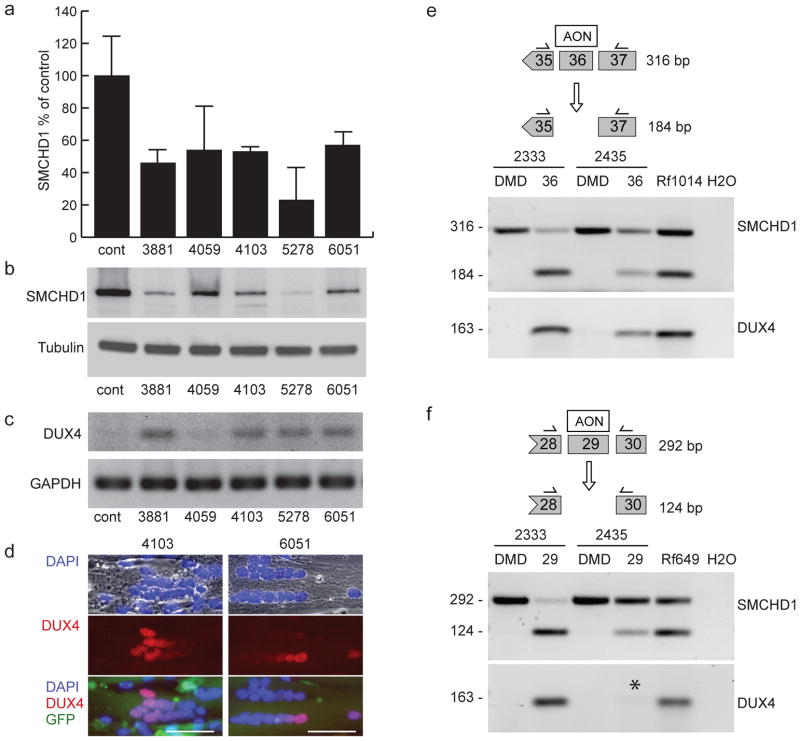
Facioscapulohumeral dystrophy (FSHD) is characterized by chromatin relaxation of the D4Z4 macrosatellite array on chromosome 4 and expression of the D4Z4-encoded DUX4 gene in skeletal muscle. The more common form, autosomal dominant FSHD1, is caused by contraction of the D4Z4 array, whereas the genetic determinants and inheritance of D4Z4 array contraction-independent FSHD2 are unclear. Here, we show that mutations in SMCHD1 (encoding structural maintenance of chromosomes flexible hinge domain containing 1) on chromosome 18 reduce SMCHD1 protein levels and segregate with genome-wide D4Z4 CpG hypomethylation in human kindreds. FSHD2 occurs in individuals who inherited both the SMCHD1 mutation and a normal-sized D4Z4 array on a chromosome 4 haplotype permissive for DUX4 expression. Reducing SMCHD1 levels in skeletal muscle results in D4Z4 contraction-independent DUX4 expression. Our study identifies SMCHD1 as an epigenetic modifier of the D4Z4 metastable epiallele and as a causal genetic determinant of FSHD2 and possibly other human diseases subject to epigenetic regulation.
Figures
References
-
- Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy: molecular pathological advances and future directions. Curr Opin Neurol. 2011;24:423–428. - PubMed
-
- de Greef JC, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–1459. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 HG006493/HG/NHGRI NIH HHS/United States
- RC2 HG005608/HG/NHGRI NIH HHS/United States
- R01 AR064197/AR/NIAMS NIH HHS/United States
- P01NS069539/NS/NINDS NIH HHS/United States
- P01 NS069539/NS/NINDS NIH HHS/United States
- HG006493/HG/NHGRI NIH HHS/United States
- R01 AR066248/AR/NIAMS NIH HHS/United States
- R01 AR045203/AR/NIAMS NIH HHS/United States
- UL1 RR024160/RR/NCRR NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- UL1RR024160/RR/NCRR NIH HHS/United States
- R01AR045203/AR/NIAMS NIH HHS/United States
- HG005608/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
