

Tumor dormancy, oncogene addiction, cellular senescence, and self-renewal programs
- PMID: 23143977
- PMCID: PMC3773491
- DOI: 10.1007/978-1-4614-1445-2_6
Tumor dormancy, oncogene addiction, cellular senescence, and self-renewal programs
Abstract
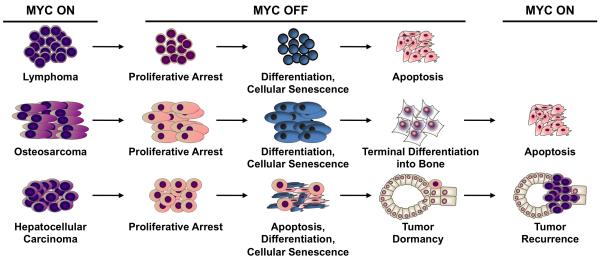
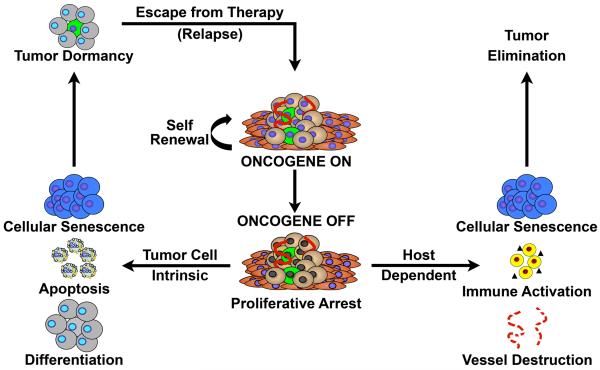
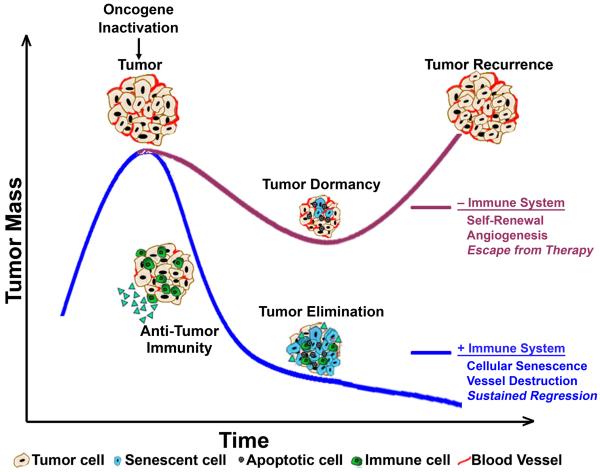
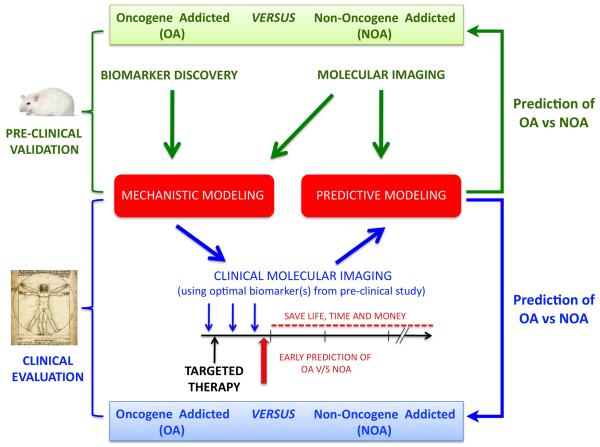
Cancers are frequently addicted to initiating oncogenes that elicit aberrant cellular proliferation, self-renewal, and apoptosis. Restoration of oncogenes to normal physiologic regulation can elicit dramatic reversal of the neoplastic phenotype, including reduced proliferation and increased apoptosis of tumor cells (Science 297(5578):63-64, 2002). In some cases, oncogene inactivation is associated with compete elimination of a tumor. However, in other cases, oncogene inactivation induces a conversion of tumor cells to a dormant state that is associated with cellular differentiation and/or loss of the ability to self-replicate. Importantly, this dormant state is reversible, with tumor cells regaining the ability to self-renew upon oncogene reactivation. Thus, understanding the mechanism of oncogene inactivation-induced dormancy may be crucial for predicting therapeutic outcome of targeted therapy. One important mechanistic insight into tumor dormancy is that oncogene addiction might involve regulation of a decision between self-renewal and cellular senescence. Recent evidence suggests that this decision is regulated by multiple mechanisms that include tumor cell-intrinsic, cell-autonomous mechanisms and host-dependent, tumor cell-non-autonomous programs (Mol Cell 4(2):199-207, 1999; Science 297(5578):102-104, 2002; Nature 431(7012):1112-1117, 2004; Proc Natl Acad Sci U S A 104(32):13028-13033, 2007). In particular, the tumor microenvironment, which is known to be critical during tumor initiation (Cancer Cell 7(5):411-423, 2005; J Clin Invest 121(6):2436-2446, 2011), prevention (Nature 410(6832):1107-1111, 2001), and progression (Cytokine Growth Factor Rev 21(1):3-10, 2010), also appears to dictate when oncogene inactivation elicits the permanent loss of self-renewal through induction of cellular senescence (Nat Rev Clin Oncol 8(3):151-160, 2011; Science 313(5795):1960-1964, 2006; N Engl J Med 351(21):2159-21569, 2004). Thus, oncogene addiction may be best modeled as a consequence of the interplay amongst cell-autonomous and host-dependent programs that define when a therapy will result in tumor dormancy.
Figures
References
-
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297(5578):63–4. - PubMed
-
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. - PubMed
-
- Jain M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102–4. - PubMed
-
- Shachaf CM, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431(7012):1112–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
