

On the mechanism of synaptic depression induced by CaMKIIN, an endogenous inhibitor of CaMKII
- PMID: 23145145
- PMCID: PMC3493544
- DOI: 10.1371/journal.pone.0049293
On the mechanism of synaptic depression induced by CaMKIIN, an endogenous inhibitor of CaMKII
Abstract
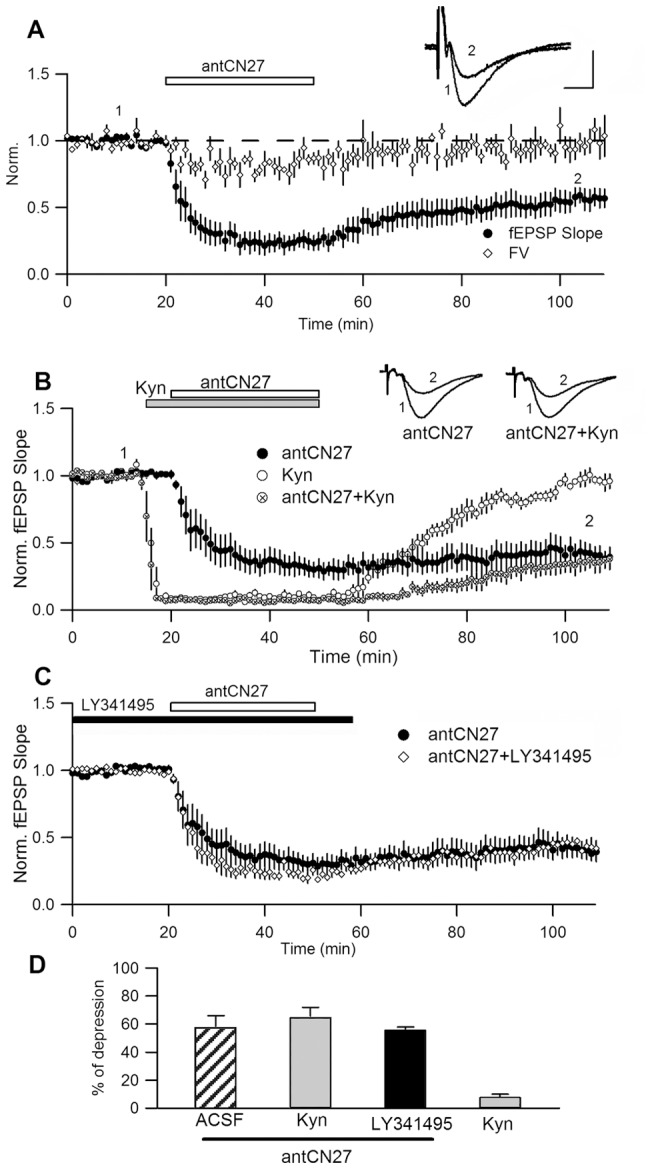
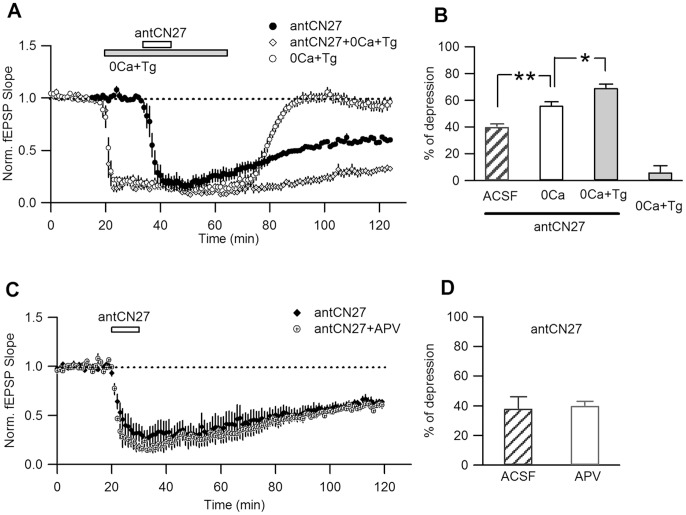
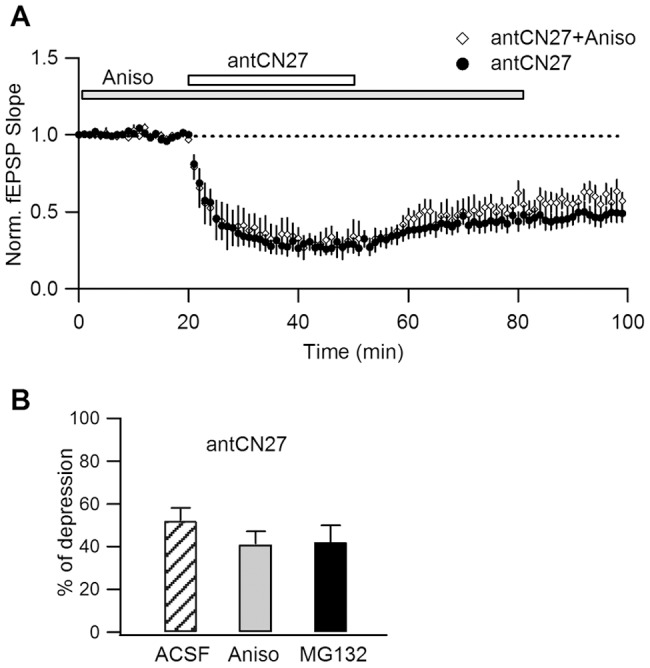
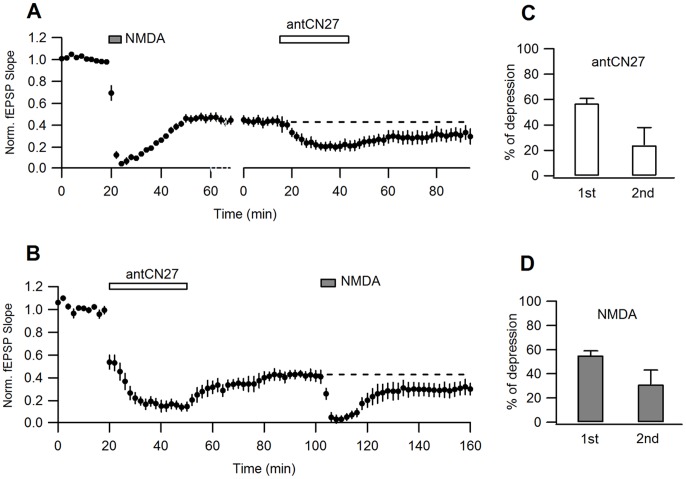
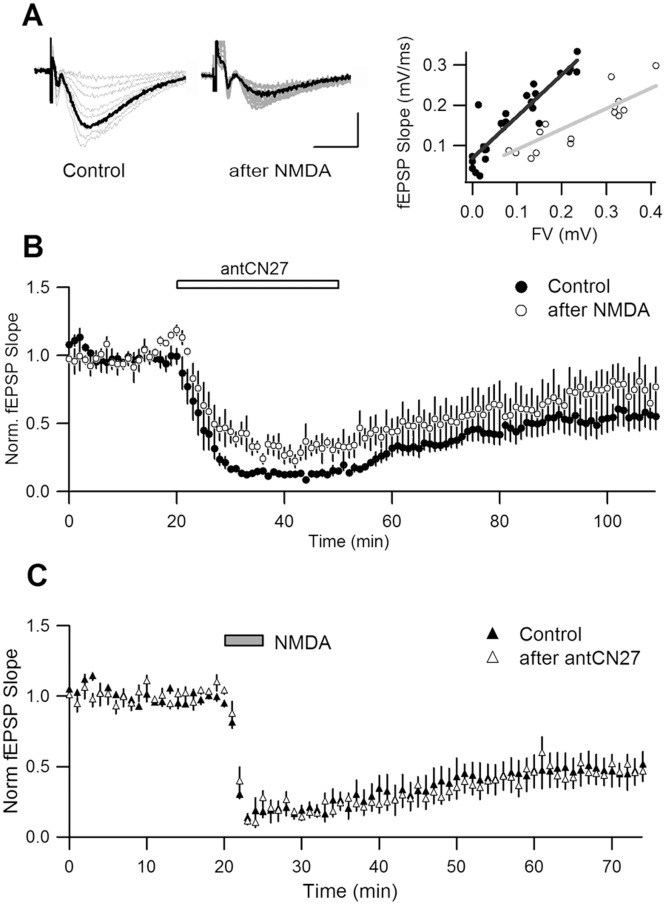
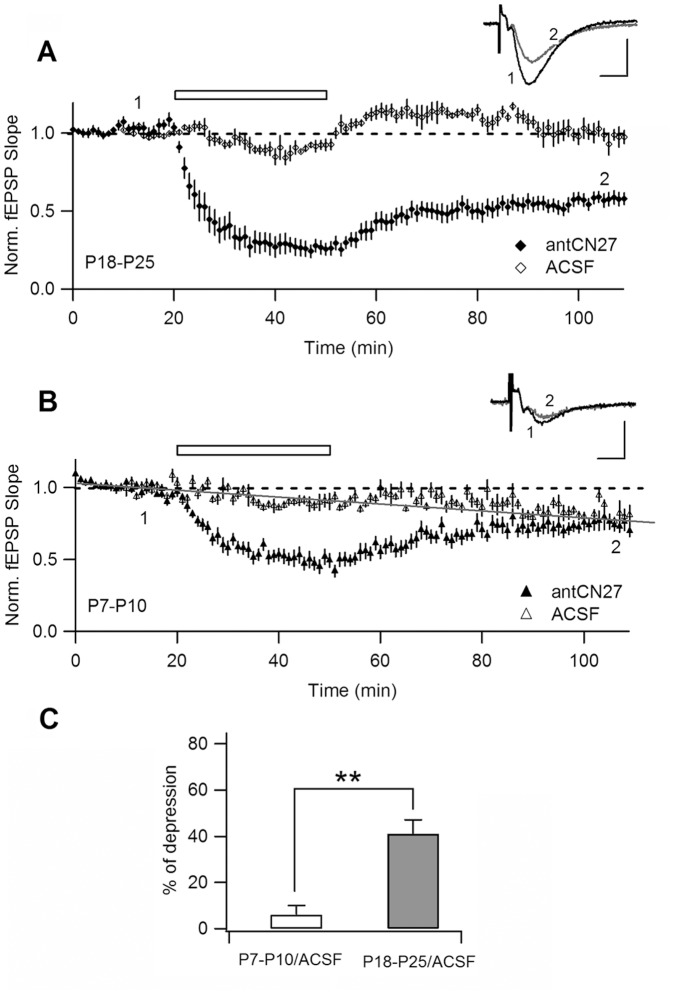
Activity-dependent synaptic plasticity underlies, at least in part, learning and memory processes. NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) is a major synaptic plasticity model. During LTP induction, Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) is activated, autophosphorylated and persistently translocated to the postsynaptic density, where it binds to the NMDAR. If any of these steps is inhibited, LTP is disrupted. The endogenous CaMKII inhibitor proteins CaMKIINα,β are rapidly upregulated in specific brain regions after learning. We recently showed that transient application of peptides derived from CaMKIINα (CN peptides) persistently depresses synaptic strength and reverses LTP saturation, as it allows further LTP induction in previously saturated pathways. The treatment disrupts basal CaMKII-NMDAR interaction and decreases bound CaMKII fraction in spines. To unravel CaMKIIN function and to further understand CaMKII role in synaptic strength maintenance, here we more deeply investigated the mechanism of synaptic depression induced by CN peptides (CN-depression) in rat hippocampal slices. We showed that CN-depression does not require glutamatergic synaptic activity or Ca(2+) signaling, thus discarding unspecific triggering of activity-dependent long-term depression (LTD) in slices. Moreover, occlusion experiments revealed that CN-depression and NMDAR-LTD have different expression mechanisms. We showed that CN-depression does not involve complex metabolic pathways including protein synthesis or proteasome-mediated degradation. Remarkably, CN-depression cannot be resolved in neonate rats, for which CaMKII is mostly cytosolic and virtually absent at the postsynaptic densities. Overall, our results support a direct effect of CN peptides on synaptic CaMKII-NMDAR binding and suggest that CaMKIINα,β could be critical plasticity-related proteins that may operate as cell-wide homeostatic regulators preventing saturation of LTP mechanisms or may selectively erase LTP-induced traces in specific groups of synapses.
Conflict of interest statement
Figures
Similar articles
-
Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength.J Neurosci. 2011 Jun 22;31(25):9170-8. doi: 10.1523/JNEUROSCI.1250-11.2011. J Neurosci. 2011. PMID: 21697368 Free PMC article.
-
Stimulating β-adrenergic receptors promotes synaptic potentiation by switching CaMKII movement from LTD to LTP mode.J Biol Chem. 2023 Jun;299(6):104706. doi: 10.1016/j.jbc.2023.104706. Epub 2023 Apr 13. J Biol Chem. 2023. PMID: 37061000 Free PMC article.
-
CaMKII T286 phosphorylation has distinct essential functions in three forms of long-term plasticity.J Biol Chem. 2022 Sep;298(9):102299. doi: 10.1016/j.jbc.2022.102299. Epub 2022 Jul 21. J Biol Chem. 2022. PMID: 35872016 Free PMC article.
-
The role of CaMKII autophosphorylation for NMDA receptor-dependent synaptic potentiation.Neuropharmacology. 2021 Aug 1;193:108616. doi: 10.1016/j.neuropharm.2021.108616. Epub 2021 May 26. Neuropharmacology. 2021. PMID: 34051268 Review.
-
CaMKII regulation in information processing and storage.Trends Neurosci. 2012 Oct;35(10):607-18. doi: 10.1016/j.tins.2012.05.003. Epub 2012 Jun 19. Trends Neurosci. 2012. PMID: 22717267 Free PMC article. Review.
Cited by
-
Synaptic memory and CaMKII.Physiol Rev. 2023 Oct 1;103(4):2877-2925. doi: 10.1152/physrev.00034.2022. Epub 2023 Jun 8. Physiol Rev. 2023. PMID: 37290118 Free PMC article. Review.
-
Criteria for identifying the molecular basis of the engram (CaMKII, PKMzeta).Mol Brain. 2017 Nov 29;10(1):55. doi: 10.1186/s13041-017-0337-4. Mol Brain. 2017. PMID: 29187215 Free PMC article. Review.
-
Heterogeneous CaMKII-Dependent Synaptic Compensations in CA1 Pyramidal Neurons From Acute Hippocampal Slices.Front Cell Neurosci. 2022 Mar 30;16:821088. doi: 10.3389/fncel.2022.821088. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35431809 Free PMC article.
-
Identification of functional tRNA-derived fragments in senescence-accelerated mouse prone 8 brain.Aging (Albany NY). 2019 Nov 20;11(22):10485-10498. doi: 10.18632/aging.102471. Epub 2019 Nov 20. Aging (Albany NY). 2019. PMID: 31746776 Free PMC article.
-
Enzymatic activity of CaMKII is not required for its interaction with the glutamate receptor subunit GluN2B.Mol Pharmacol. 2013 Dec;84(6):834-43. doi: 10.1124/mol.113.089045. Epub 2013 Sep 20. Mol Pharmacol. 2013. PMID: 24056996 Free PMC article.
References
-
- Colbran RJ, Brown AM (2004) Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol 14: 318–327. - PubMed
-
- Strack S, Choi S, Lovinger DM, Colbran RJ (1997) Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem 272: 13467–13470. - PubMed
-
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization. Science 284: 162–166. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
