

Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study
- PMID: 23147228
- PMCID: PMC3551719
- DOI: 10.1186/1750-1172-7-88
Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study
Abstract
Background: Due partly to physicians' unawareness, many adults with Pompe disease are diagnosed with great delay. Besides, it is not well known which factors influence the rate of disease progression, and thus disease outcome. We delineated the specific clinical features of Pompe disease in adults, and mapped out the distribution and severity of muscle weakness, and the sequence of involvement of the individual muscle groups. Furthermore, we defined the natural disease course and identified prognostic factors for disease progression.
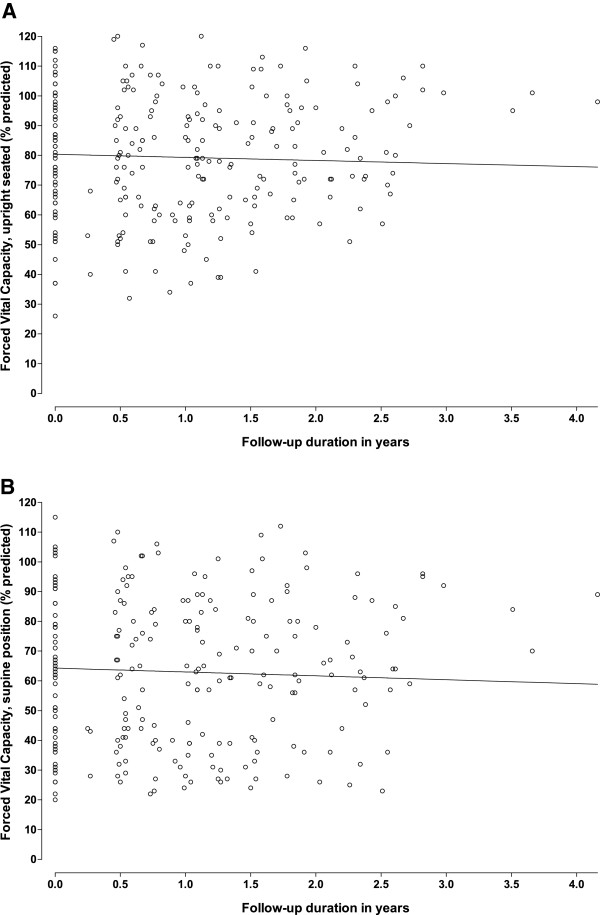
Methods: We conducted a single-center, prospective, observational study. Muscle strength (manual muscle testing, and hand-held dynamometry), muscle function (quick motor function test), and pulmonary function (forced vital capacity in sitting and supine positions) were assessed every 3-6 months and analyzed using repeated-measures ANOVA.
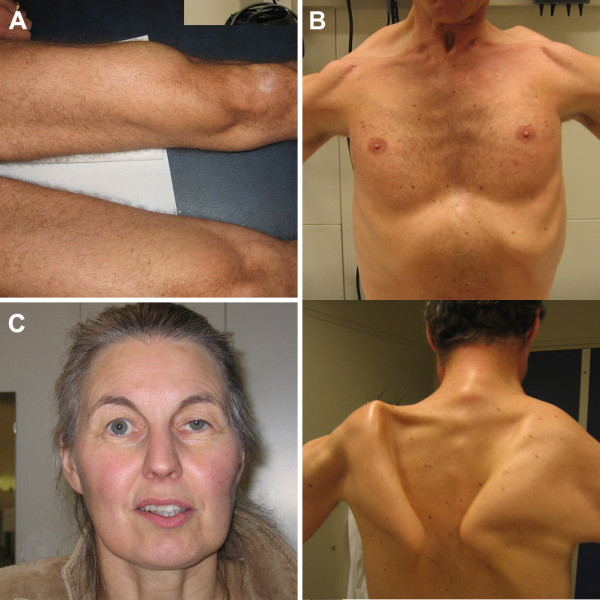
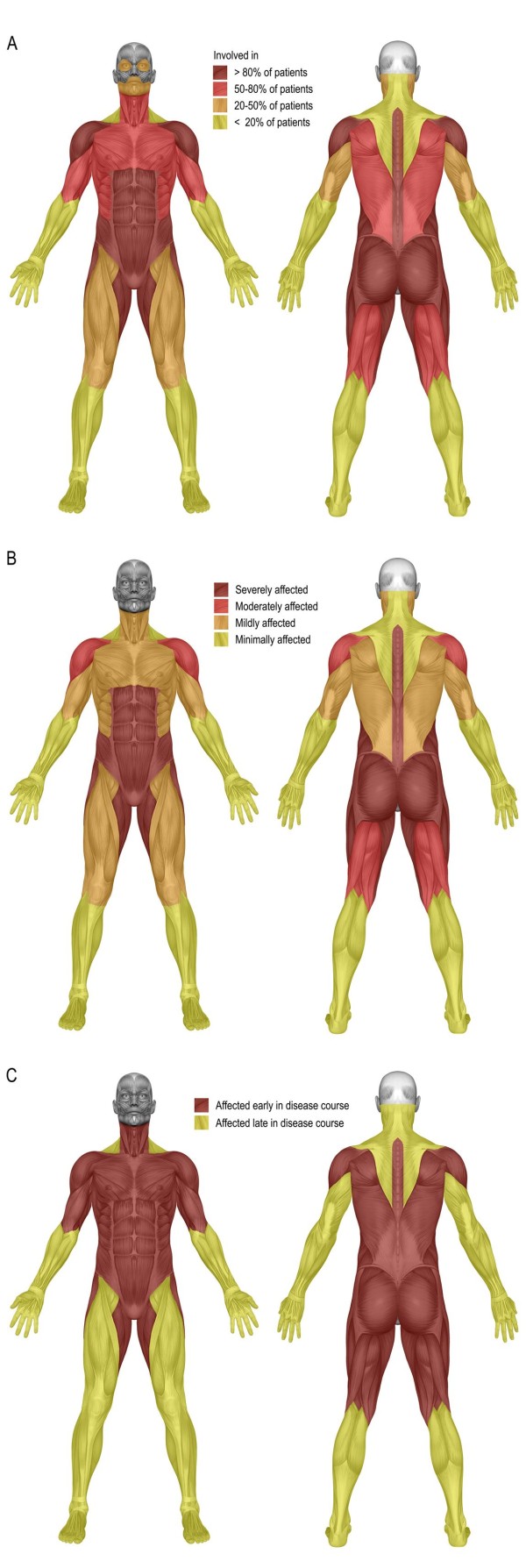
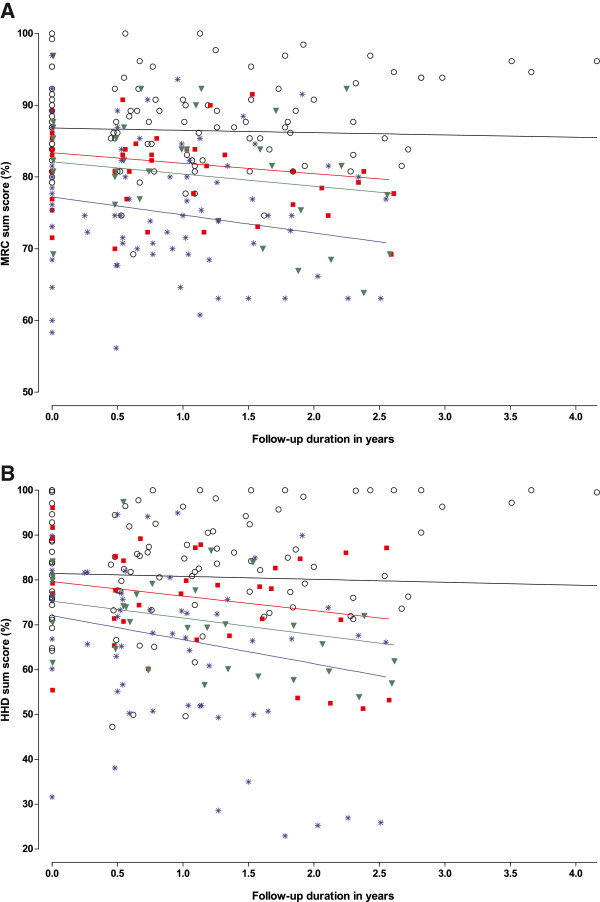
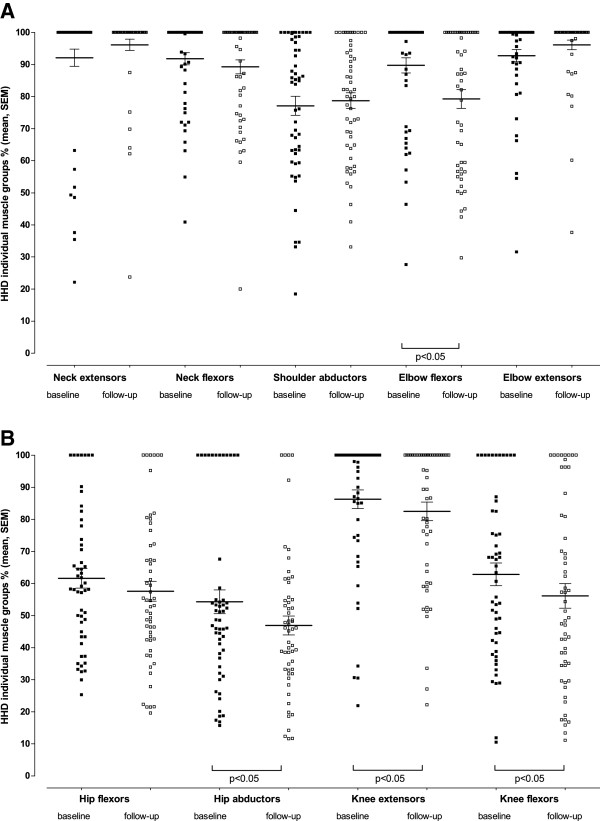
Results: Between October 2004 and August 2009, 94 patients aged between 25 and 75 years were included in the study. Although skeletal muscle weakness was typically distributed in a limb-girdle pattern, many patients had unfamiliar features such as ptosis (23%), bulbar weakness (28%), and scapular winging (33%). During follow-up (average 1.6 years, range 0.5-4.2 years), skeletal muscle strength deteriorated significantly (mean declines of -1.3% point/year for manual muscle testing and of -2.6% points/year for hand-held dynamometry; both p<0.001). Longer disease duration (>15 years) and pulmonary involvement (forced vital capacity in sitting position <80%) at study entry predicted faster decline. On average, forced vital capacity in supine position deteriorated by 1.3% points per year (p=0.02). Decline in pulmonary function was consistent across subgroups. Ten percent of patients declined unexpectedly fast.
Conclusions: Recognizing patterns of common and less familiar characteristics in adults with Pompe disease facilitates timely diagnosis. Longer disease duration and reduced pulmonary function stand out as predictors of rapid disease progression, and aid in deciding whether to initiate enzyme replacement therapy, or when.
Figures
References
-
- Pompe JC. Over idiopathische hypertrofie van het hart. Ned Tijdschr Geneeskd. 1932;76:304–311.
-
- Engel AG. Franzini-Armstrong: Myology. New York: McGraw-Hill; 2004.
-
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen sto rage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
