

Toward detection of DNA-bound proteins using solid-state nanopores: insights from computer simulations
- PMID: 23147918
- PMCID: PMC3789251
- DOI: 10.1002/elps.201200164
Toward detection of DNA-bound proteins using solid-state nanopores: insights from computer simulations
Abstract
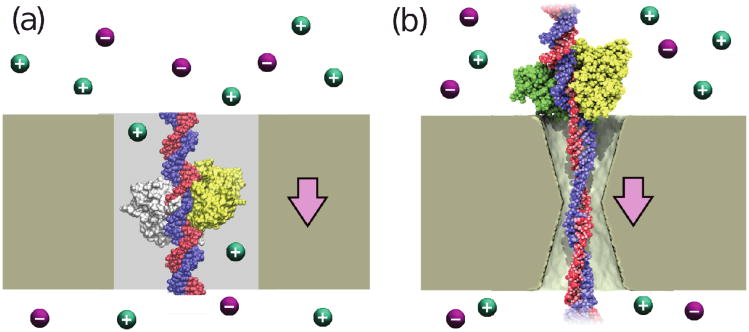
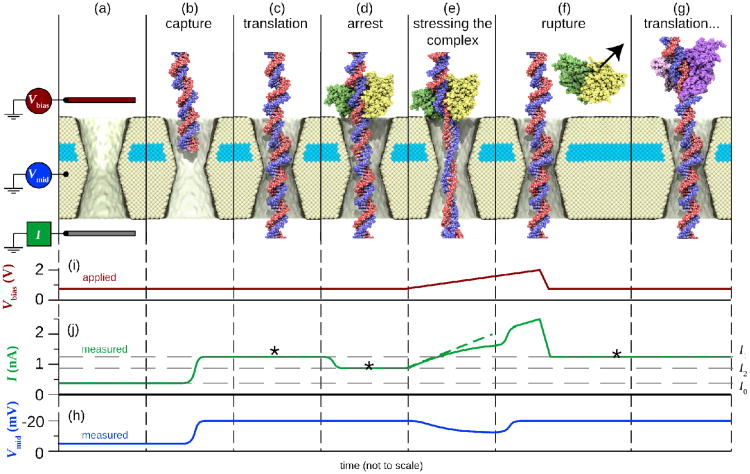
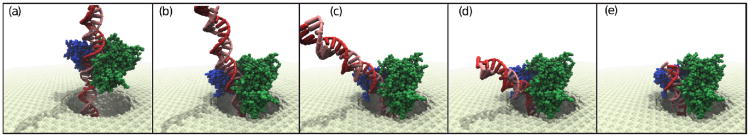
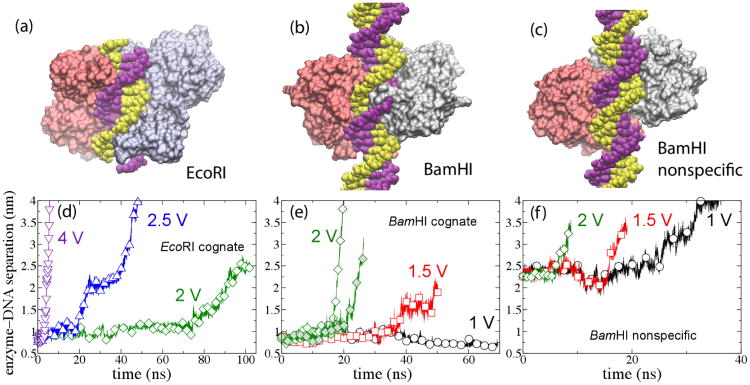
Through all-atom molecular dynamics simulations, we explore the use of nanopores in thin synthetic membranes for detection and identification of DNA binding proteins. Reproducing the setup of a typical experiment, we simulate electric field driven transport of DNA-bound proteins through nanopores smaller in diameter than the proteins. As model systems, we use restriction enzymes EcoRI and BamHI specifically and nonspecifically bound to a fragment of dsDNA, and streptavidin and NeutrAvidin proteins bound to dsDNA and ssDNA via a biotin linker. Our simulations elucidate the molecular mechanics of nanopore-induced rupture of a protein-DNA complex, the effective force applied to the DNA-protein bond by the electrophoretic force in a nanopore, and the role of DNA-surface interactions in the rupture process. We evaluate the ability of the nanopore ionic current and the local electrostatic potential measured by an embedded electrode to report capture of DNA, capture of a DNA-bound protein, and rupture of the DNA-protein bond. We find that changes in the strain on dsDNA can reveal the rupture of a protein-DNA complex by altering both the nanopore ionic current and the potential of the embedded electrode. Based on the results of our simulations, we suggest a new method for detection of DNA binding proteins that utilizes peeling of a nicked double strand under the electrophoretic force in a nanopore.
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
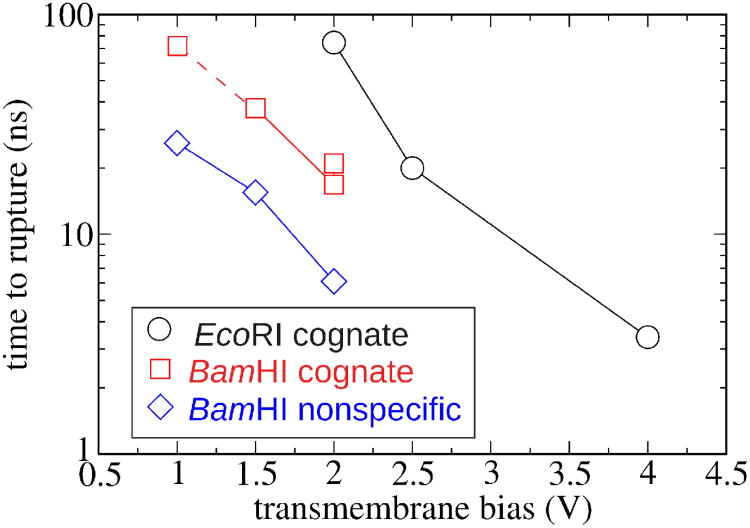
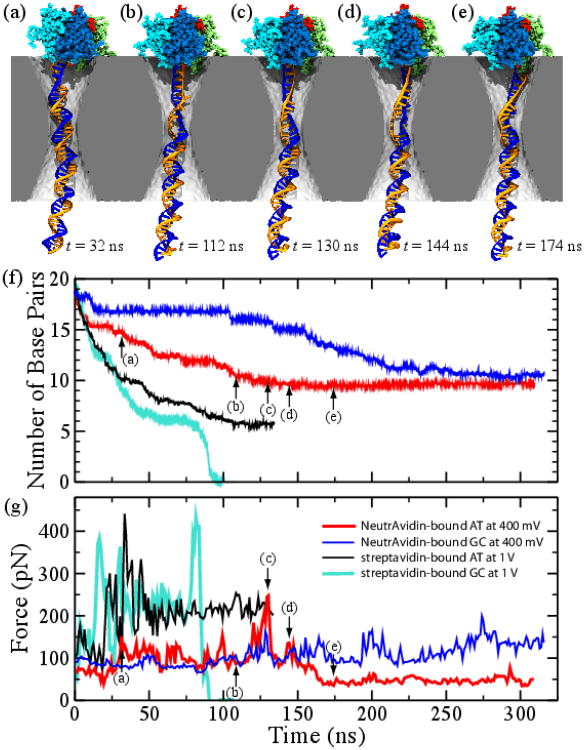
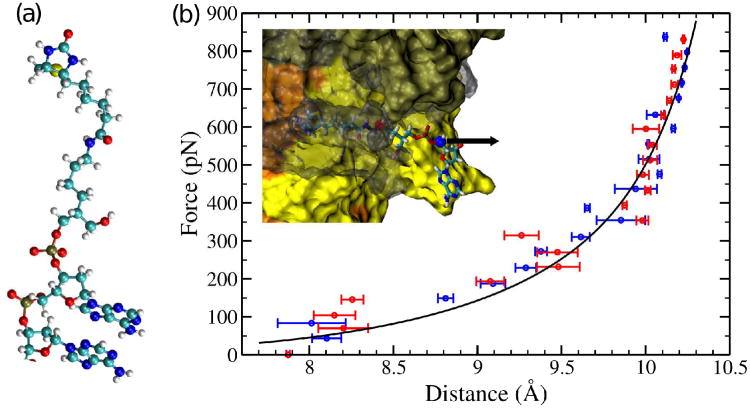
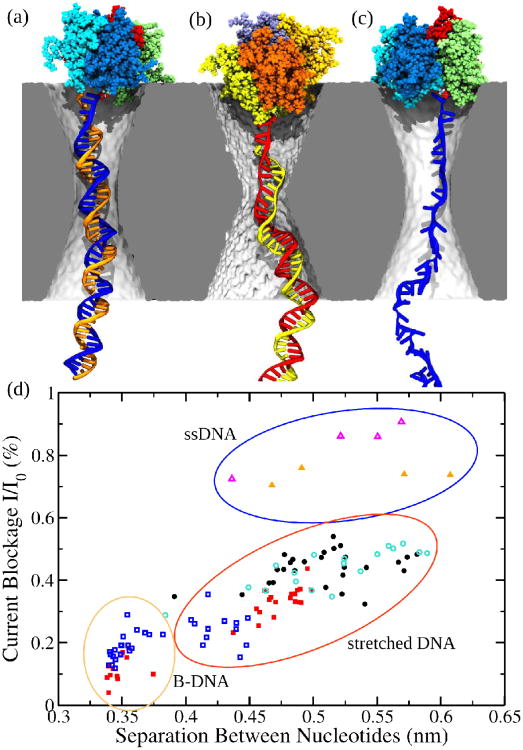
Similar articles
-
Analyzing the forces binding a restriction endonuclease to DNA using a synthetic nanopore.Nucleic Acids Res. 2009 Jul;37(12):4170-9. doi: 10.1093/nar/gkp317. Epub 2009 May 11. Nucleic Acids Res. 2009. PMID: 19433506 Free PMC article.
-
Identifying the Location of a Single Protein along the DNA Strand Using Solid-State Nanopores.ACS Nano. 2015 May 26;9(5):5289-98. doi: 10.1021/acsnano.5b00784. Epub 2015 May 6. ACS Nano. 2015. PMID: 25938865
-
Mechanical Trapping of DNA in a Double-Nanopore System.Nano Lett. 2016 Dec 14;16(12):8021-8028. doi: 10.1021/acs.nanolett.6b04642. Epub 2016 Dec 1. Nano Lett. 2016. PMID: 27960493 Free PMC article.
-
Multi-resolution simulation of DNA transport through large synthetic nanostructures.Phys Chem Chem Phys. 2022 Feb 2;24(5):2706-2716. doi: 10.1039/d1cp04589j. Phys Chem Chem Phys. 2022. PMID: 35050282 Free PMC article. Review.
-
Probing nanopores: molecular dynamics insights into the mechanisms of DNA and protein translocation through solid-state and biological nanopores.Soft Matter. 2025 Mar 26;21(13):2385-2399. doi: 10.1039/d4sm01534g. Soft Matter. 2025. PMID: 40094904 Review.
Cited by
-
Molecular Dynamics Simulation of DNA Capture and Transport in Heated Nanopores.ACS Appl Mater Interfaces. 2016 May 25;8(20):12599-608. doi: 10.1021/acsami.6b00463. Epub 2016 Mar 21. ACS Appl Mater Interfaces. 2016. PMID: 26963065 Free PMC article.
-
Molecular Determinants of Current Blockade Produced by Peptide Transport Through a Nanopore.ACS Nanosci Au. 2023 Nov 14;4(1):21-29. doi: 10.1021/acsnanoscienceau.3c00046. eCollection 2024 Feb 21. ACS Nanosci Au. 2023. PMID: 38406313 Free PMC article. Review.
-
Graphene Nanopores for Protein Sequencing.Adv Funct Mater. 2016 Jul 19;26(27):4830-4838. doi: 10.1002/adfm.201601272. Epub 2016 Jun 9. Adv Funct Mater. 2016. PMID: 27746710 Free PMC article.
-
Dynamics of a Molecular Plug Docked onto a Solid-State Nanopore.J Phys Chem Lett. 2018 Aug 16;9(16):4686-4694. doi: 10.1021/acs.jpclett.8b01755. Epub 2018 Aug 3. J Phys Chem Lett. 2018. PMID: 30058336 Free PMC article.
-
Fast, label-free force spectroscopy of histone-DNA interactions in individual nucleosomes using nanopores.J Am Chem Soc. 2013 Oct 16;135(41):15350-2. doi: 10.1021/ja408354s. Epub 2013 Oct 2. J Am Chem Soc. 2013. PMID: 24079416 Free PMC article.
References
-
- Li J, Stein D, McMullan C, Branton D, Aziz MJ, Golovchenko JA. Nature. 2001;412:166–169. - PubMed
-
- Dekker C. Nature Nanotech. 2007;2:209–215. - PubMed
-
- Storm AJ, Chen JH, Zandbergen HW, Dekker C. Phys Rev E. 2005;71:051903–051913. - PubMed
-
- Skinner GM, van den Hout M, Broekmans O, Dekker C, Dekker NH. Nano Lett. 2009;9:2953–2960. - PubMed
-
- Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Nature Nanotech. 2010;5:807–814. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
