

OXPHOS mutations and neurodegeneration
- PMID: 23149385
- PMCID: PMC3545297
- DOI: 10.1038/emboj.2012.300
OXPHOS mutations and neurodegeneration
Abstract
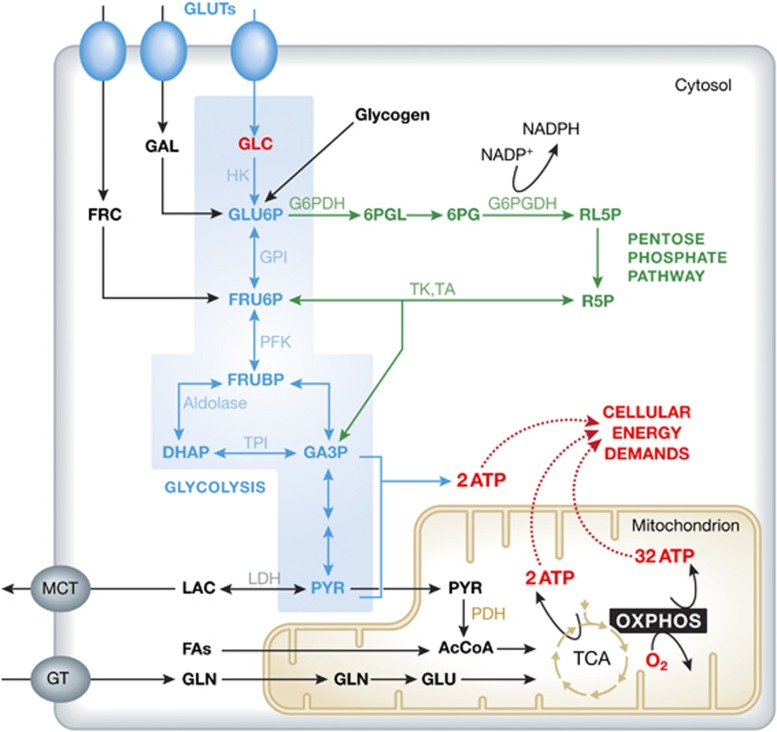
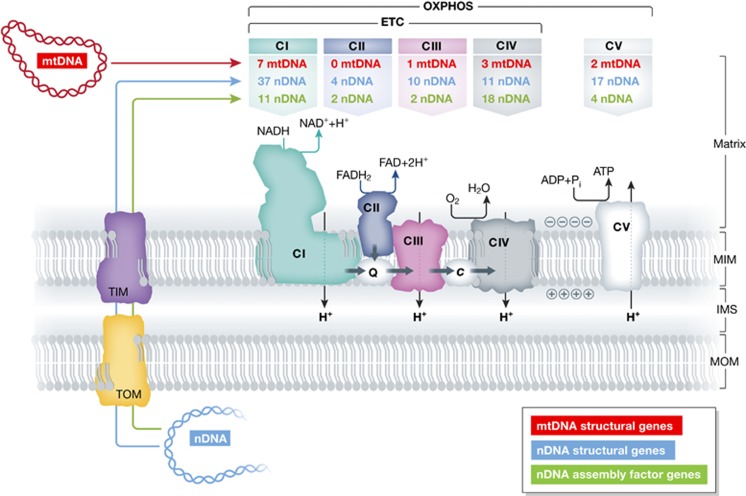
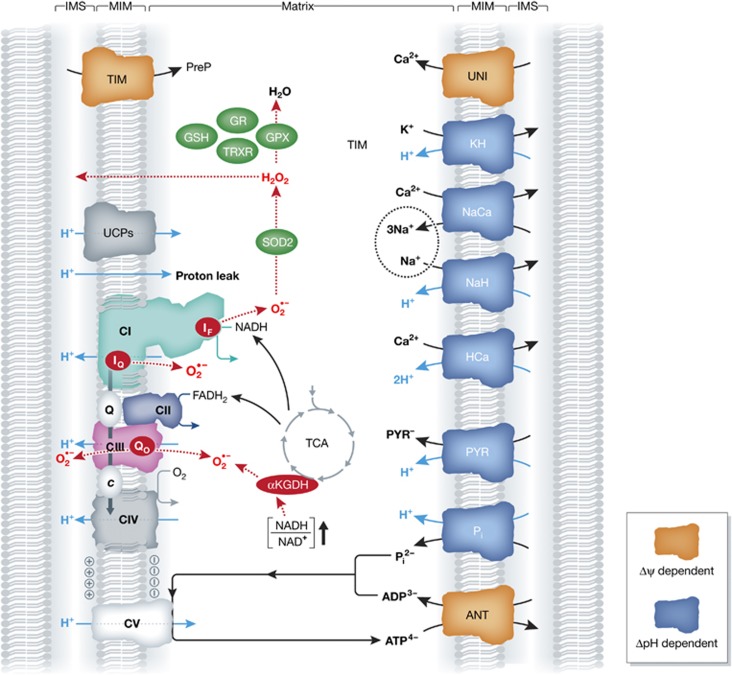
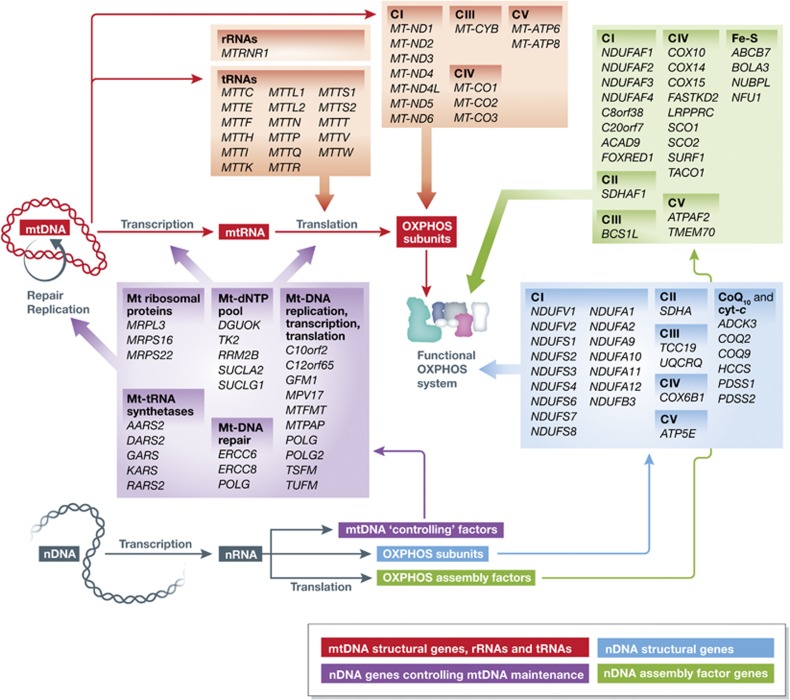
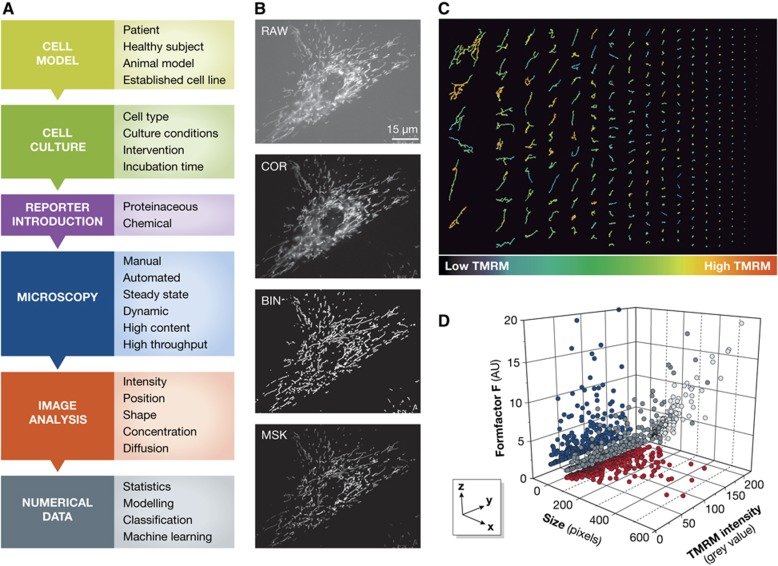
Mitochondrial oxidative phosphorylation (OXPHOS) sustains organelle function and plays a central role in cellular energy metabolism. The OXPHOS system consists of 5 multisubunit complexes (CI-CV) that are built up of 92 different structural proteins encoded by the nuclear (nDNA) and mitochondrial DNA (mtDNA). Biogenesis of a functional OXPHOS system further requires the assistance of nDNA-encoded OXPHOS assembly factors, of which 35 are currently identified. In humans, mutations in both structural and assembly genes and in genes involved in mtDNA maintenance, replication, transcription, and translation induce 'primary' OXPHOS disorders that are associated with neurodegenerative diseases including Leigh syndrome (LS), which is probably the most classical OXPHOS disease during early childhood. Here, we present the current insights regarding function, biogenesis, regulation, and supramolecular architecture of the OXPHOS system, as well as its genetic origin. Next, we provide an inventory of OXPHOS structural and assembly genes which, when mutated, induce human neurodegenerative disorders. Finally, we discuss the consequences of mutations in OXPHOS structural and assembly genes at the single cell level and how this information has advanced our understanding of the role of OXPHOS dysfunction in neurodegeneration.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA (2008) Respiratory active mitochondrial supercomplexes. Mol Cell 32: 529–539 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
