

Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane
- PMID: 23150667
- PMCID: PMC3543034
- DOI: 10.1074/jbc.M112.428573
Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane
Abstract
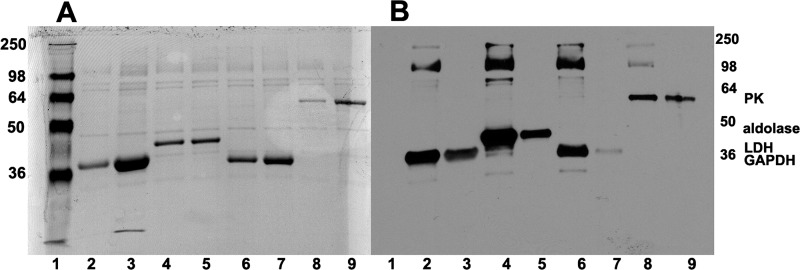
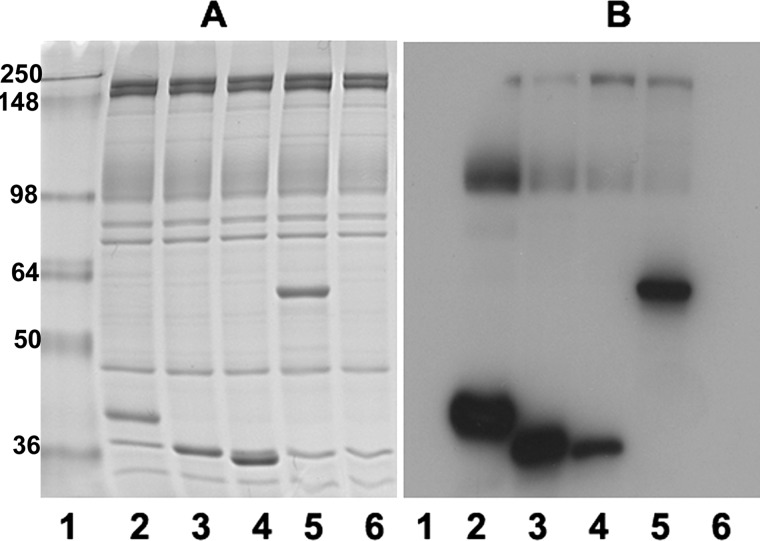
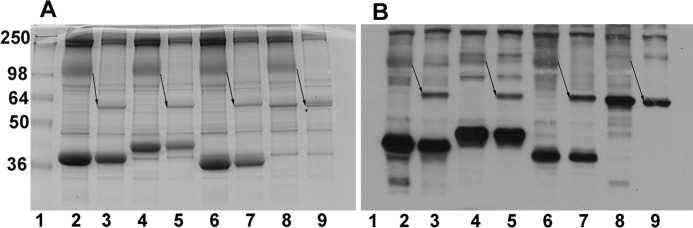
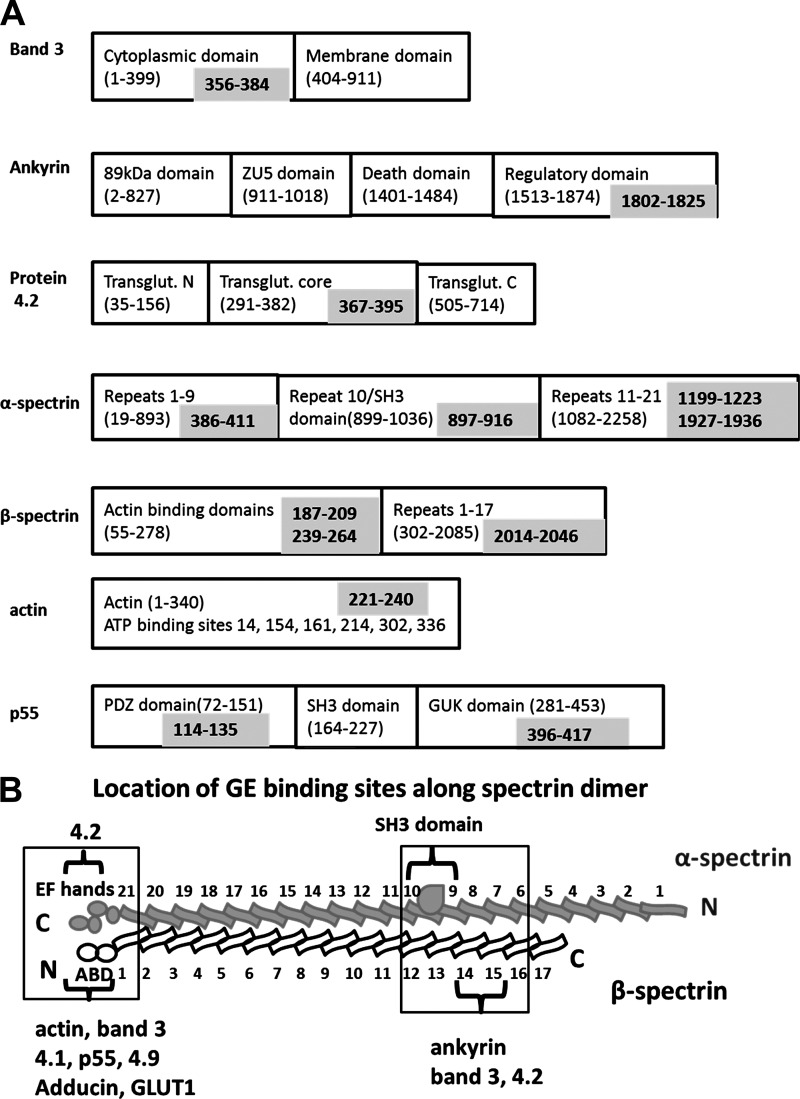
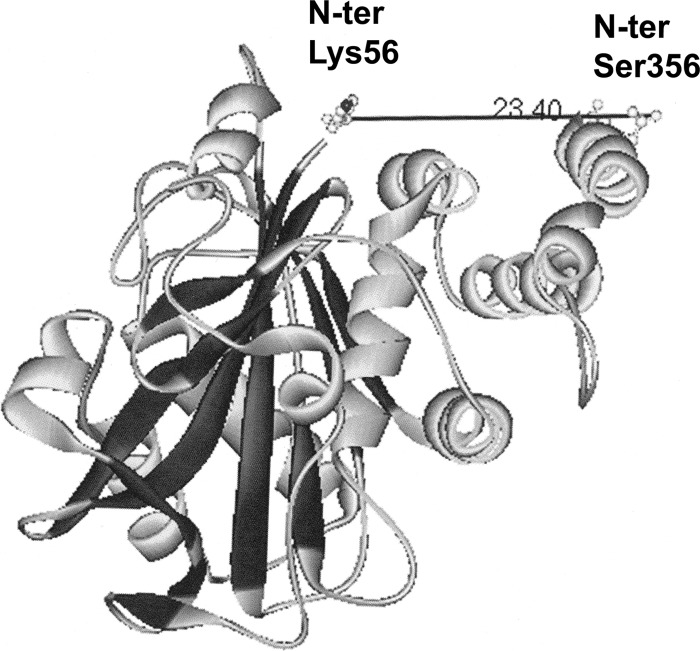
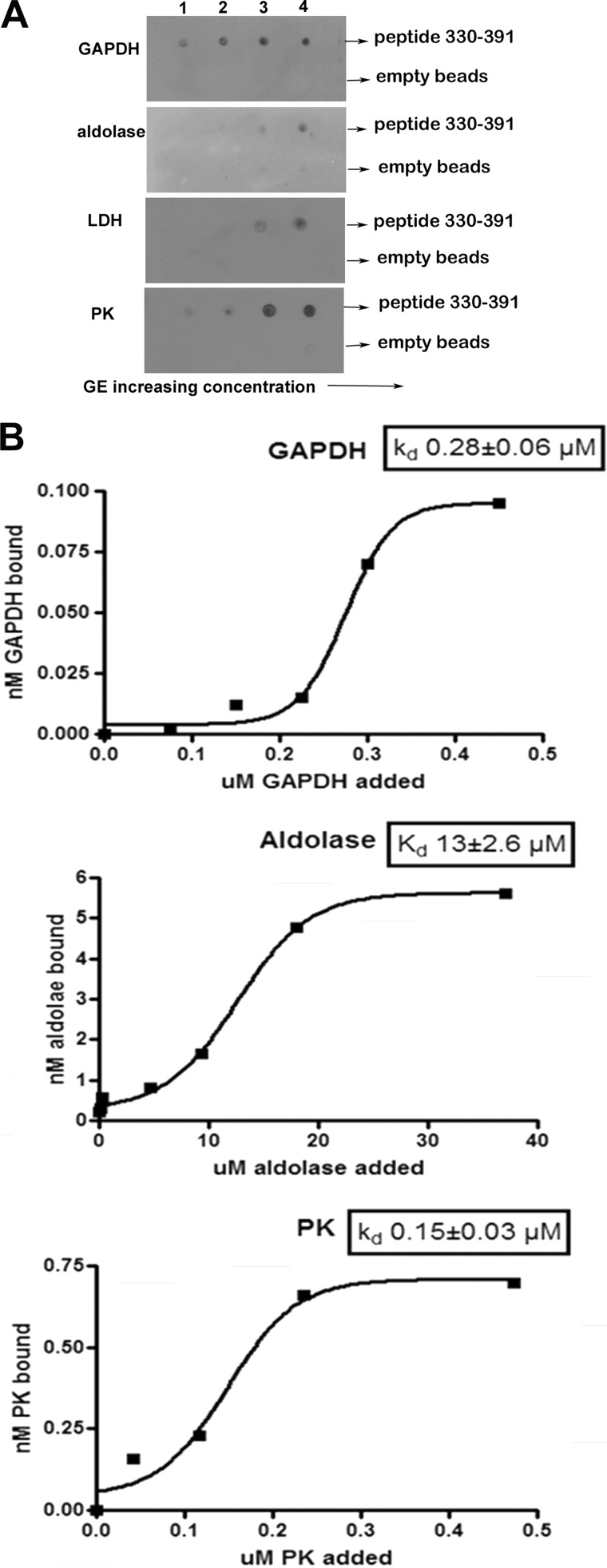
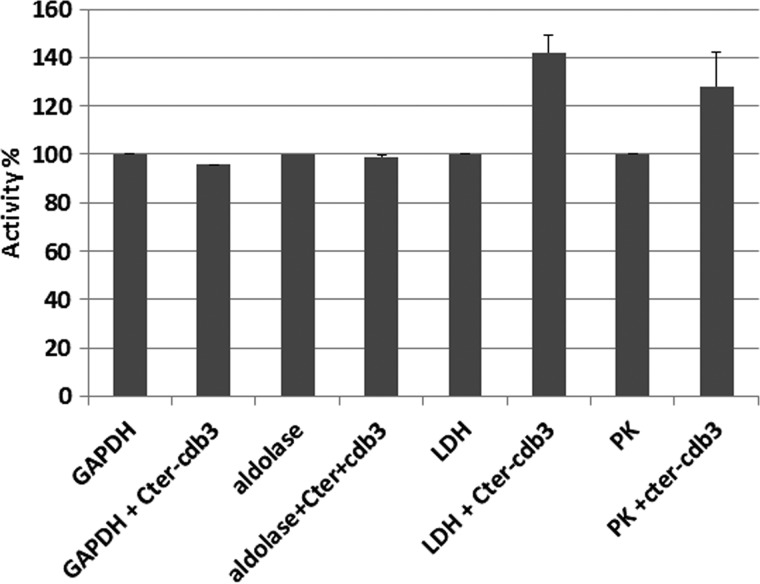
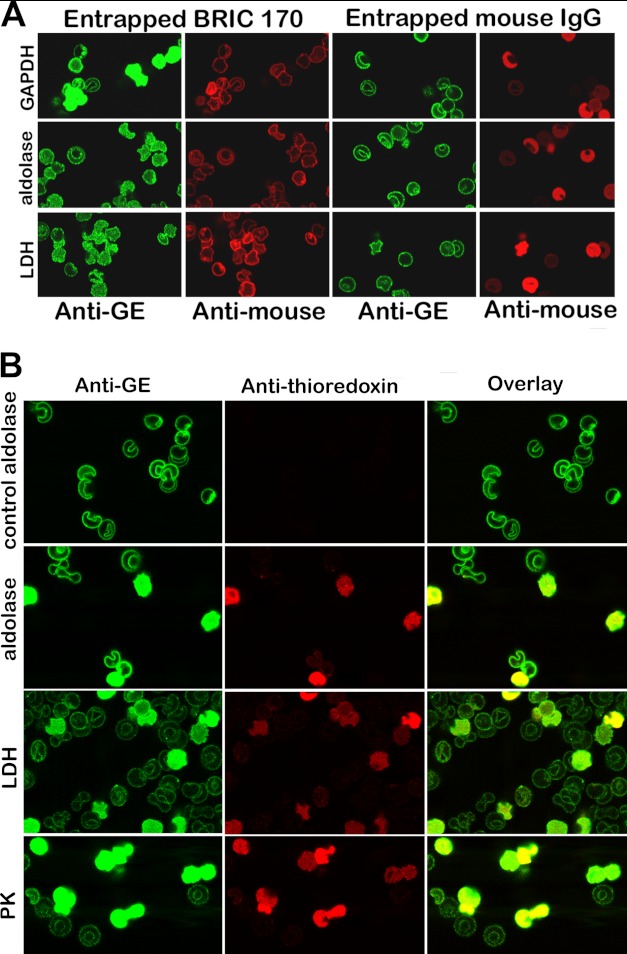
Glycolytic enzymes (GEs) have been shown to exist in multienzyme complexes on the inner surface of the human erythrocyte membrane. Because no protein other than band 3 has been found to interact with GEs, and because several GEs do not bind band 3, we decided to identify the additional membrane proteins that serve as docking sites for GE on the membrane. For this purpose, a method known as "label transfer" that employs a photoactivatable trifunctional cross-linking reagent to deliver a biotin from a derivatized GE to its binding partner on the membrane was used. Mass spectrometry analysis of membrane proteins that were biotinylated following rebinding and photoactivation of labeled GAPDH, aldolase, lactate dehydrogenase, and pyruvate kinase revealed not only the anticipated binding partner, band 3, but also the association of GEs with specific peptides in α- and β-spectrin, ankyrin, actin, p55, and protein 4.2. More importantly, the labeled GEs were also found to transfer biotin to other GEs in the complex, demonstrating for the first time that GEs also associate with each other in their membrane complexes. Surprisingly, a new GE binding site was repeatedly identified near the junction of the membrane-spanning and cytoplasmic domains of band 3, and this binding site was confirmed by direct binding studies. These results not only identify new components of the membrane-associated GE complexes but also provide molecular details on the specific peptides that form the interfacial contacts within each interaction.
Figures
References
-
- Vonck J., Schäfer E. (2009) Supramolecular organization of protein complexes in the mitochondrial inner membrane. Biochim. Biophys. Acta 1793, 117–124 - PubMed
-
- Sterling D., Reithmeier R. A., Casey J. R. (2001) A transport metabolon. J. Biol. Chem. 276, 47886–47894 - PubMed
-
- Sowah D., Casey J. R. (2011) An intramolecular transport metabolon. Fusion of carbonic anhydrase II to the COOH terminus of the Cl−/HCO3− exchanger, AE1. Am. J. Physiol. Cell Physiol. 301, C336–C346 - PubMed
-
- Endeward V., Cartron J.-P., Ripoche P., Gros G. (2008) RhAG protein of the Rhesus complex is a CO2 channel in the human red cell membrane. FASEB J. 22, 64–73 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
