

C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity
- PMID: 23150673
- PMCID: PMC3537064
- DOI: 10.1074/jbc.M112.400168
C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity
Abstract
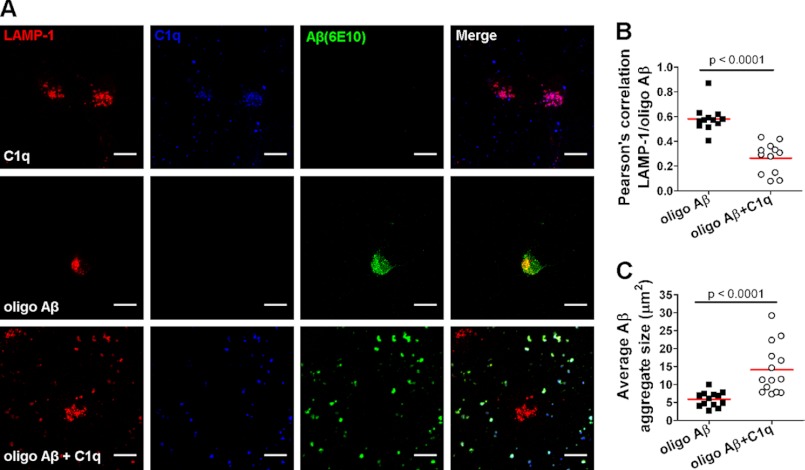
Complement protein C1q is induced in the brain in response to a variety of neuronal injuries, including Alzheimer disease (AD), and blocks fibrillar amyloid-β (fAβ) neurotoxicity in vitro. Here, we show that C1q protects immature and mature primary neurons against fAβ toxicity, and we report for the first time that C1q prevents toxicity induced by oligomeric forms of amyloid-β (Aβ). Gene expression analysis reveals C1q-activated phosphorylated cAMP-response element-binding protein and AP-1, two transcription factors associated with neuronal survival and neurite outgrowth, and increased LRP1B and G protein-coupled receptor 6(GPR6) expression in fAβ-injured neurons. Silencing of cAMP-response element-binding protein, LRP1B or GPR6 expression inhibited C1q-mediated neuroprotection from fAβ-induced injury. In addition, C1q altered the association of oligomeric Aβ and fAβ with neurons. In vivo, increased hippocampal expression of C1q, LRP1B, and GPR6 is observed as early as 2 months of age in the 3 × Tg mouse model of AD, whereas no such induction of LRP1B and GPR6 was seen in C1q-deficient AD mice. In contrast, expression of C1r and C1s, proteases required to activate the classical complement pathway, and C3 showed a significant age-dependent increase only after 10-13 months of age when Aβ plaques start to accumulate in this AD model. Thus, our results identify pathways by which C1q, up-regulated in vivo early in response to injury without the coordinate induction of other complement components, can induce a program of gene expression that promotes neuroprotection and thus may provide protection against Aβ in preclinical stages of AD and other neurodegenerative processes.
Figures







References
-
- Bohlson S. S., Fraser D. A., Tenner A. J. (2007) Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol. Immunol. 44, 33–43 - PubMed
-
- Melchior B., Garcia A. E., Hsiung B. K., Lo K. M., Doose J. M., Thrash J. C., Stalder A. K., Staufenbiel M., Neumann H., Carson M. J. (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice. Implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2, e00037. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
