

Apoptosis and necrosis: two different outcomes of cigarette smoke condensate-induced endothelial cell death
- PMID: 23152060
- PMCID: PMC3542598
- DOI: 10.1038/cddis.2012.162
Apoptosis and necrosis: two different outcomes of cigarette smoke condensate-induced endothelial cell death
Abstract
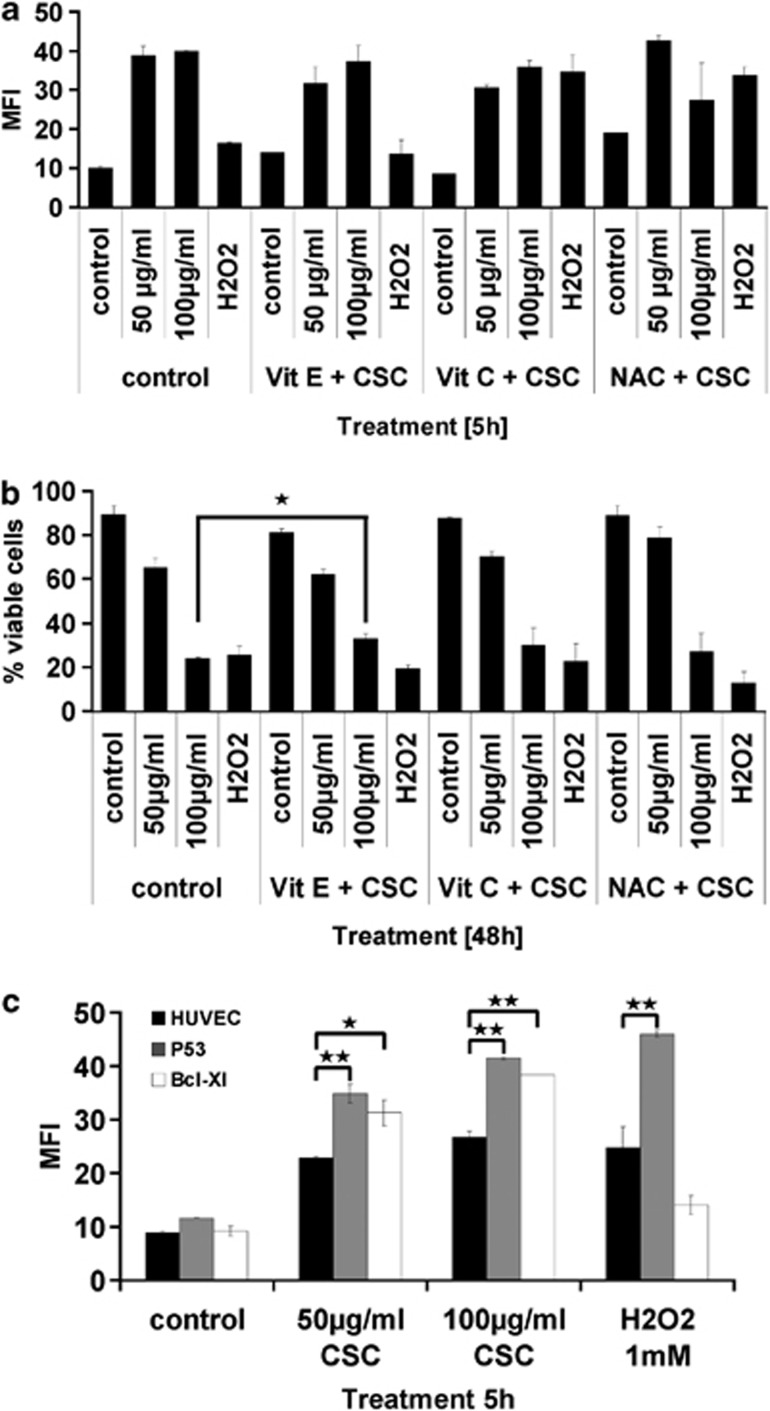
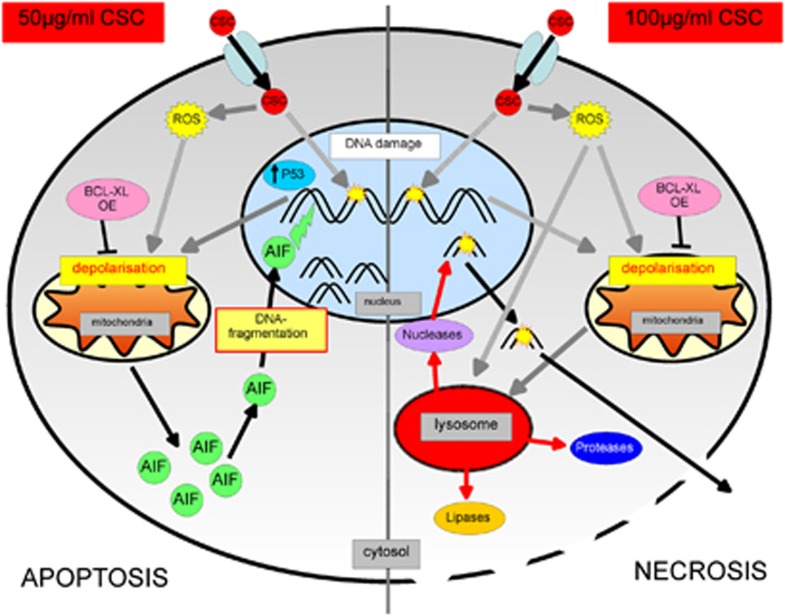
Cigarette smoking is one of the most important and preventable risk factors for atherosclerosis. However, because of the complex composition of cigarette smoke, the detailed pathophysiological mechanisms are not fully understood. Based on controversial reports on the pro-atherogenic activity of cigarette smoke condensate, also called tar fraction (CSC), we decided to analyse the effects of CSC on the viability of endothelial cells in vitro. The results of this study show that low concentrations of the hydrophobic tar fraction induces DNA damage resulting in a P53-dependent and BCL-XL-inhibitable death cascade. Western blot analyses showed that this cascade is caspase-independent and immunofluorescence analysis have shown that the apoptotic death signalling is mediated by the release of apoptosis-inducing factor. Higher CSC concentrations also induce apoptotic-like signalling but the signalling cascade is then redirected to necrosis. Despite the fact that CSC induces a profound increase in cellular reactive oxygen species production, antioxidants exhibit only a minimal cell death protective effect. Our data indicates that not only hydrophilic constituents of cigarette smoke extract, but also CSC is harmful to endothelial cells. The mode and the outcome of CSC-induced cell death signalling are highly concentration dependent: lower concentrations induce caspase-independent apoptosis-like cell death, whereas incubation with higher concentrations interrupts apoptotic signalling and induces necrosis.
Figures



References
-
- Organisation WH . Tobacco – Fact Sheet No 339. World Health Organisation; 2012.
-
- Knoflach M, Kiechl S, Kind M, Said M, Sief R, Gisinger M, et al. Cardiovascular risk factors and atherosclerosis in young males: ARMY study (atherosclerosis risk-factors in male youngsters) Circulation. 2003;108:1064–1069. - PubMed
-
- Powell JT. Vascular damage from smoking: disease mechanisms at the arterial wall. Vasc Med. 1998;3:21–28. - PubMed
-
- Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
