

Novel mutations in gB and gH circumvent the requirement for known gD Receptors in herpes simplex virus 1 entry and cell-to-cell spread
- PMID: 23152509
- PMCID: PMC3554156
- DOI: 10.1128/JVI.02804-12
Novel mutations in gB and gH circumvent the requirement for known gD Receptors in herpes simplex virus 1 entry and cell-to-cell spread
Abstract
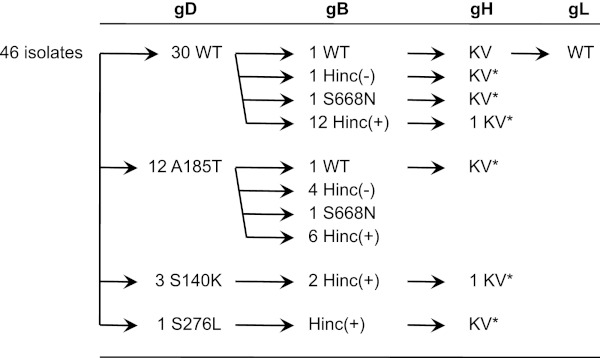
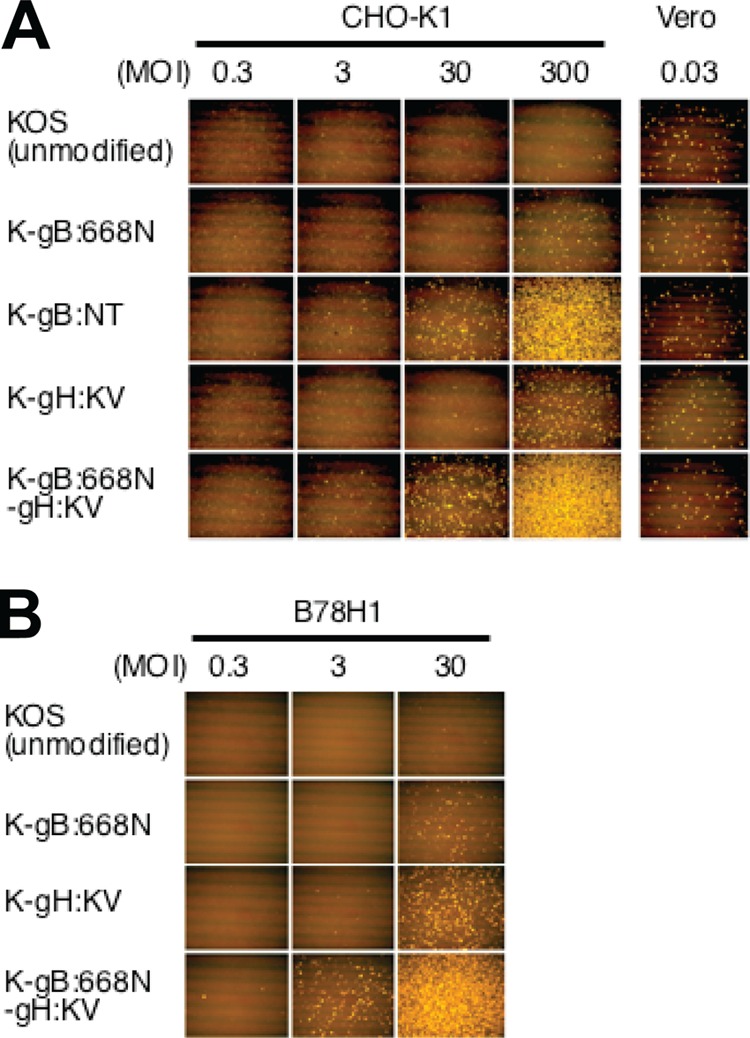
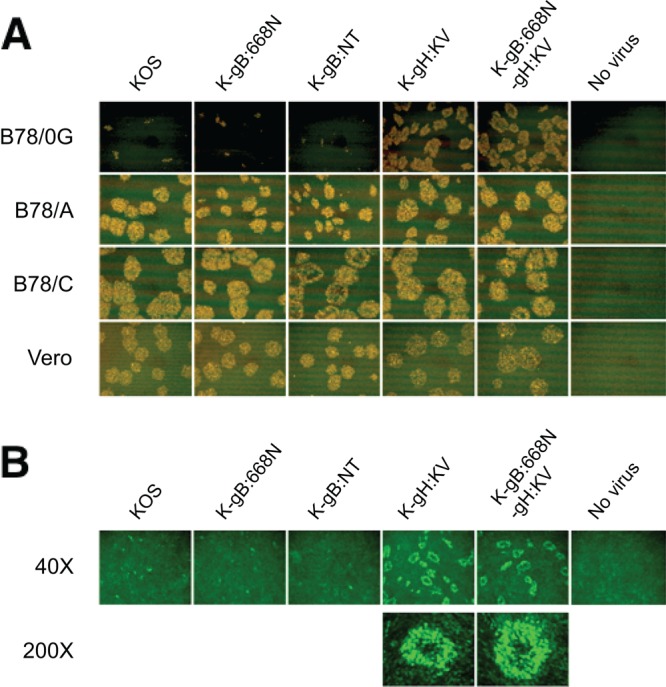
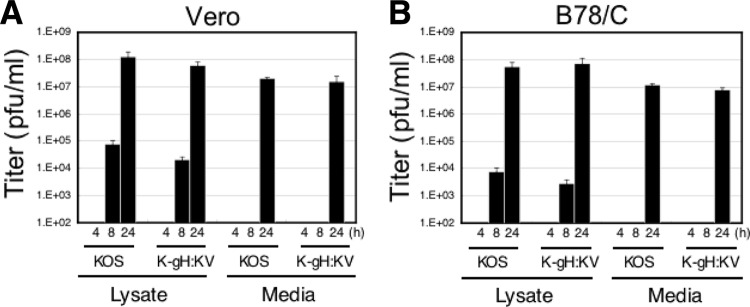
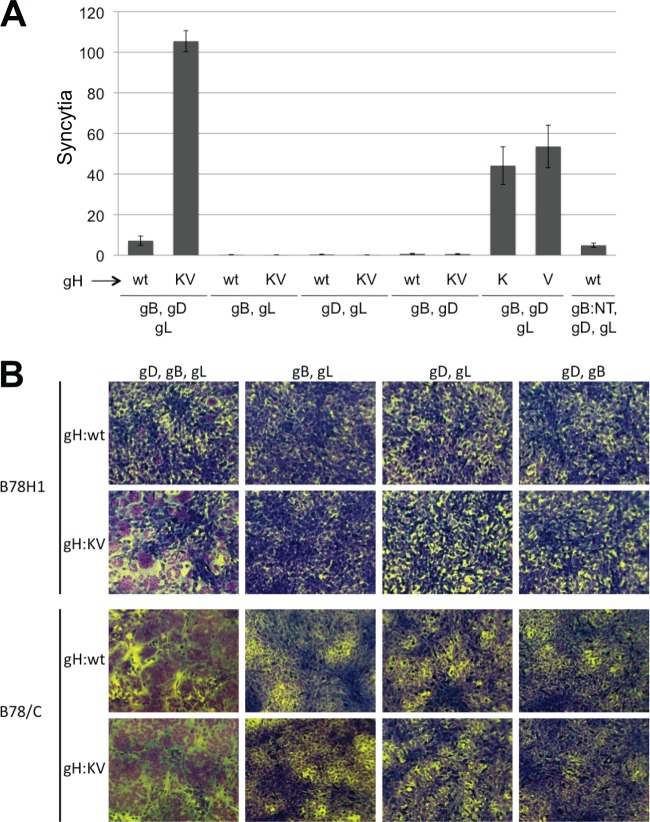
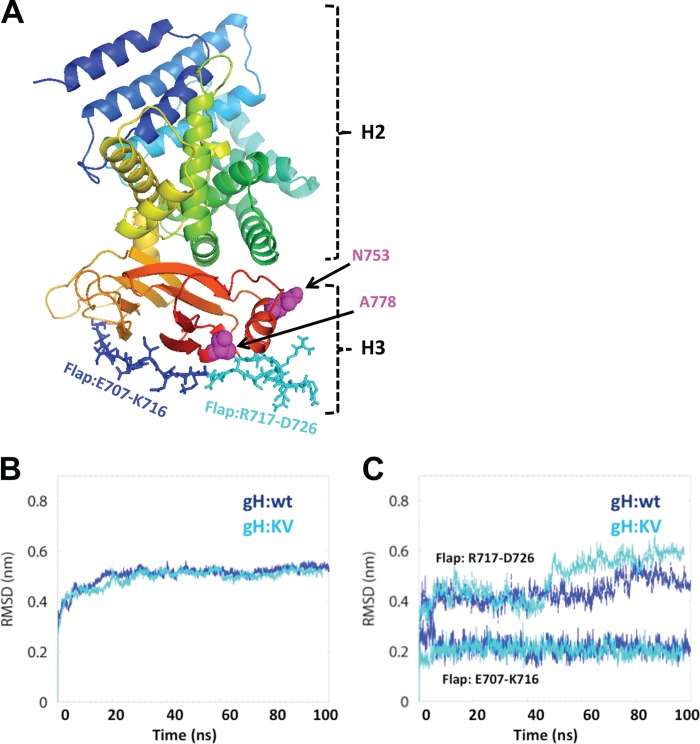
Both entry and cell-to-cell spread of herpes simplex virus (HSV) involve a cascade of cooperative interactions among the essential glycoproteins D, B, and H/L (gD, gB, and gH/gL, respectively) initiated by the binding of gD to a cognate HSV entry receptor. We previously reported that a variant (D285N/A549T) of glycoprotein B (gB:NT) enabled primary virus entry into cells that were devoid of typical HSV entry receptors. Here, we compared the activities of the gB:NT variant with those of a newly selected variant of glycoprotein H (gH:KV) and a frequently coselected gB variant (gB:S668N). In combination, gH:KV and gB:S668N enabled primary virus entry into cells that lacked established HSV entry receptors as efficiently as did gB:NT, but separately, each variant enabled only limited entry. Remarkably, gH:KV uniquely facilitated secondary virus spread between cells that lacked canonical entry receptors. Transient expression of the four essential entry glycoproteins revealed that gH:KV, but not gB:NT, induced fusion between cells lacking the standard receptors. Because the involvement of gD remained essential for virus spread and cell fusion, we propose that gH:KV mimics a transition state of gH that responds efficiently to weak signals from gD to reach the active state. Computational modeling of the structures of wild-type gH and gH:KV revealed relatively subtle differences that may have accounted for our experimental findings. Our study shows that (i) the dependence of HSV-1 entry and spread on specific gD receptors can be reduced by sequence changes in the downstream effectors gB and gH, and (ii) the relative roles of gB and gH are different in entry and spread.
Figures
Similar articles
-
Syncytial Mutations Do Not Impair the Specificity of Entry and Spread of a Glycoprotein D Receptor-Retargeted Herpes Simplex Virus.J Virol. 2016 Nov 28;90(24):11096-11105. doi: 10.1128/JVI.01456-16. Print 2016 Dec 15. J Virol. 2016. PMID: 27707922 Free PMC article.
-
Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors.mBio. 2013 Feb 26;4(2):e00046-13. doi: 10.1128/mBio.00046-13. mBio. 2013. PMID: 23443004 Free PMC article.
-
Functional Characterization of Glycoprotein H Chimeras Composed of Conserved Domains of the Pseudorabies Virus and Herpes Simplex Virus 1 Homologs.J Virol. 2015 Oct 21;90(1):421-32. doi: 10.1128/JVI.01985-15. Print 2016 Jan 1. J Virol. 2015. PMID: 26491153 Free PMC article.
-
Two Sides to Every Story: Herpes Simplex Type-1 Viral Glycoproteins gB, gD, gH/gL, gK, and Cellular Receptors Function as Key Players in Membrane Fusion.Viruses. 2021 Sep 16;13(9):1849. doi: 10.3390/v13091849. Viruses. 2021. PMID: 34578430 Free PMC article. Review.
-
Understanding HSV-1 entry glycoproteins.Rev Med Virol. 2007 May-Jun;17(3):205-15. doi: 10.1002/rmv.531. Rev Med Virol. 2007. PMID: 17295428 Free PMC article. Review.
Cited by
-
Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV.Mol Ther. 2015 Jan;23(1):99-107. doi: 10.1038/mt.2014.177. Epub 2014 Sep 9. Mol Ther. 2015. PMID: 25200130 Free PMC article.
-
Syncytial Mutations Do Not Impair the Specificity of Entry and Spread of a Glycoprotein D Receptor-Retargeted Herpes Simplex Virus.J Virol. 2016 Nov 28;90(24):11096-11105. doi: 10.1128/JVI.01456-16. Print 2016 Dec 15. J Virol. 2016. PMID: 27707922 Free PMC article.
-
Development of an oncolytic HSV vector fully retargeted specifically to cellular EpCAM for virus entry and cell-to-cell spread.Gene Ther. 2016 Jun;23(6):479-88. doi: 10.1038/gt.2016.17. Epub 2016 Feb 23. Gene Ther. 2016. PMID: 26905369
-
Dual Ligand Insertion in gB and gD of Oncolytic Herpes Simplex Viruses for Retargeting to a Producer Vero Cell Line and to Cancer Cells.J Virol. 2018 Feb 26;92(6):e02122-17. doi: 10.1128/JVI.02122-17. Print 2018 Mar 15. J Virol. 2018. PMID: 29263257 Free PMC article.
-
Functions of the UL51 protein during the herpesvirus life cycle.Front Microbiol. 2024 Aug 26;15:1457582. doi: 10.3389/fmicb.2024.1457582. eCollection 2024. Front Microbiol. 2024. PMID: 39252835 Free PMC article. Review.
References
-
- Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J. Gen. Virol. 69:1147–1156 - PubMed
-
- Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. 1998. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 72:6119–6130 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
