

Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver
- PMID: 23152807
- PMCID: PMC3494720
- DOI: 10.1371/journal.pone.0048801
Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver
Abstract
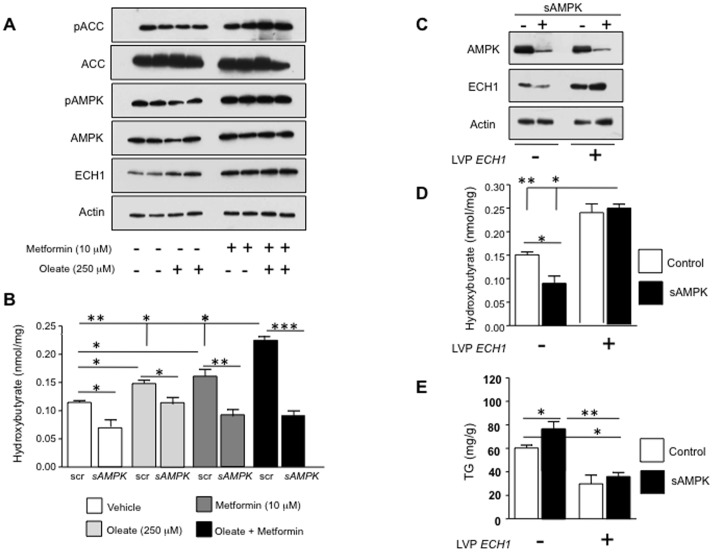
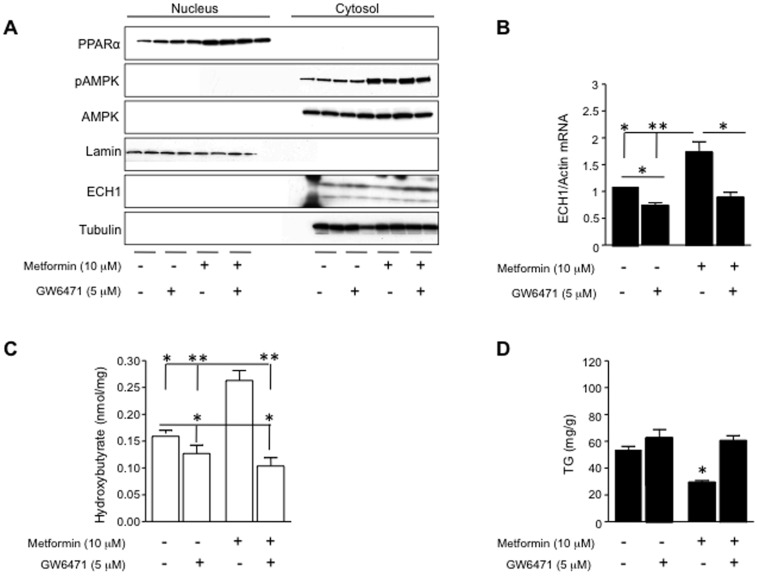
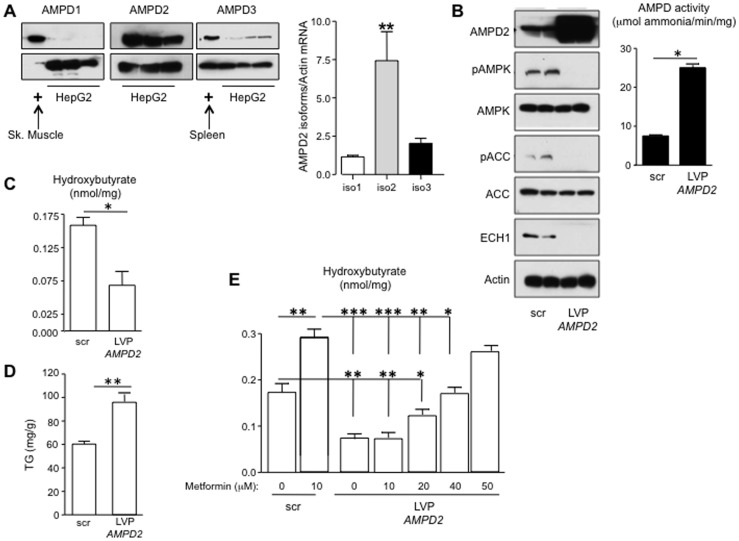
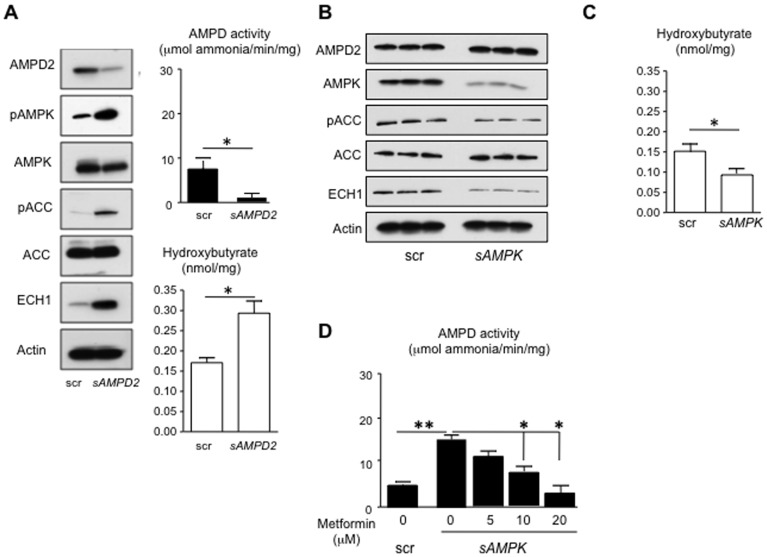
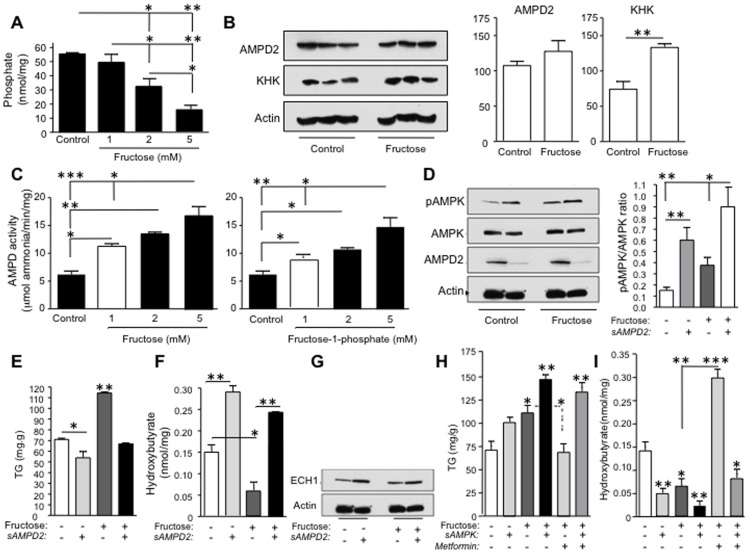
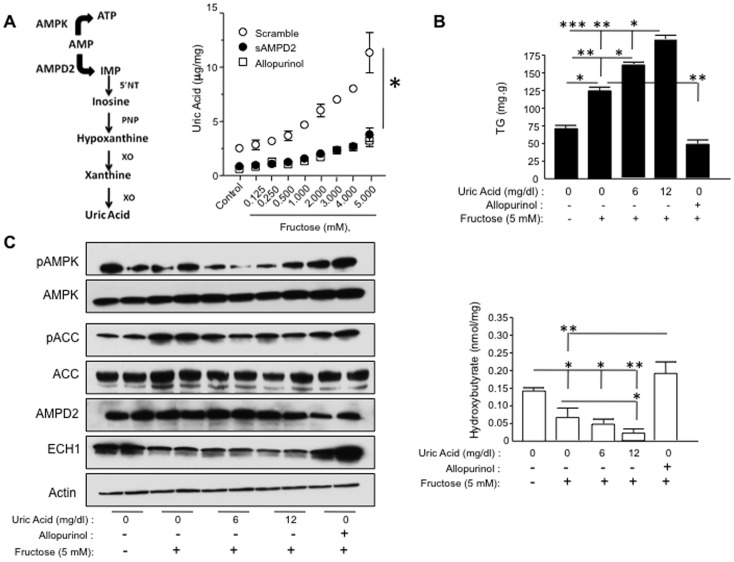
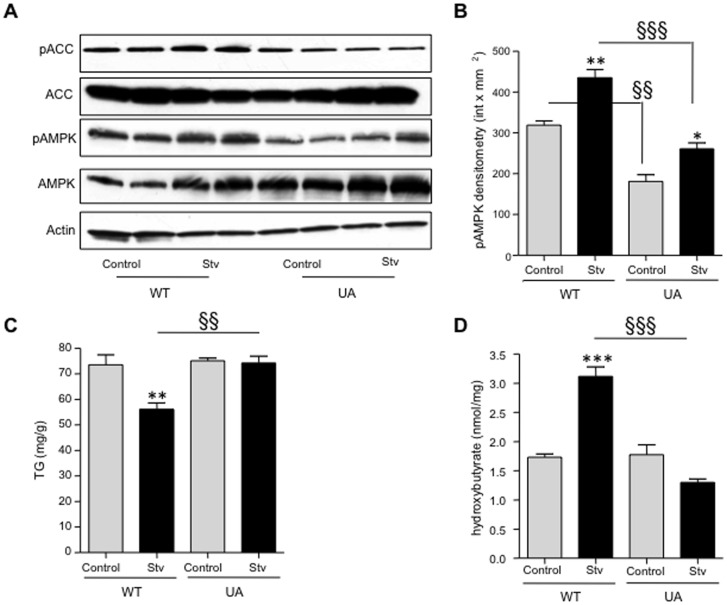
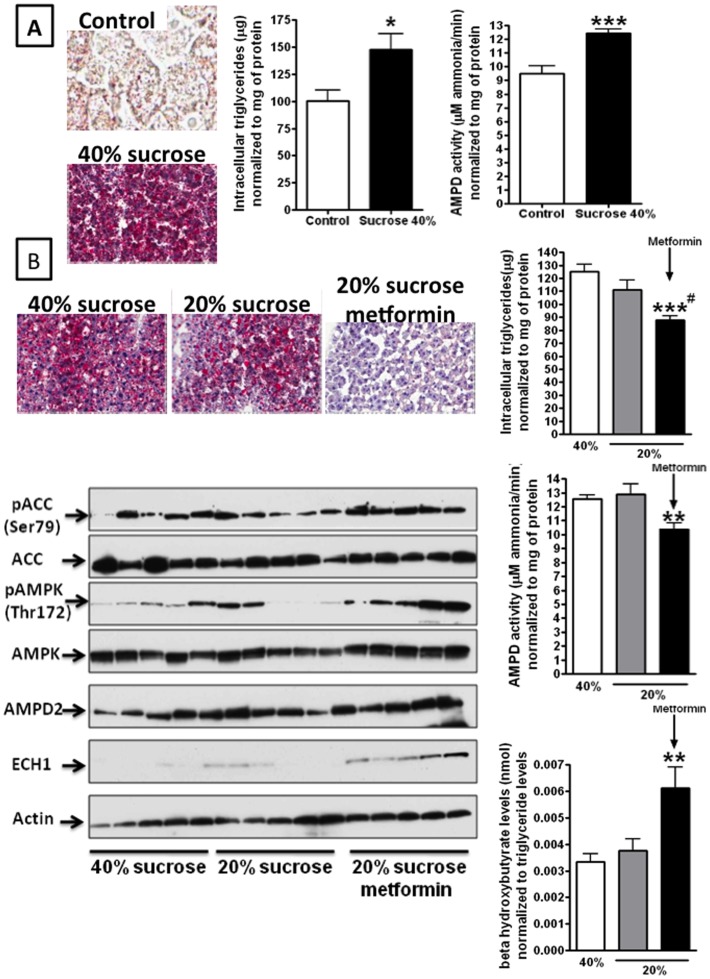
Fatty liver (hepatic steatosis) is associated with nucleotide turnover, loss of ATP and generation of adenosine monophosphate (AMP). It is well known that in fatty liver, activity of the AMP-activated kinase (AMPK) is reduced and that its stimulation can prevent hepatic steatosis by both enhancing fat oxidation and reducing lipogenesis. Here we show that another AMP dependent enzyme, AMPD2, has opposing effects on fatty acid oxidation when compared to AMPK. In human hepatocytres, AMPD2 activation -either by overexpression or by lowering intracellular phosphate levels with fructose- is associated with a significant reduction in AMPK activity. Likewise, silencing of AMPK spontaneously increases AMPD activity, demonstrating that these enzymes counter-regulate each other. Furthermore, we show that a downstream product of AMP metabolism through AMPD2, uric acid, can inhibit AMPK activity in human hepatocytes. Finally, we show that fructose-induced fat accumulation in hepatocytes is due to a dominant stimulation of AMPD2 despite stimulating AMPK. In this regard, AMPD2-deficient hepatocytes demonstrate a further activation of AMPK after fructose exposure in association with increased fatty acid oxidation, and conversely silencing AMPK enhances AMPD-dependent fat accumulation. In vivo, we show that sucrose fed rats also develop fatty liver that is blocked by metformin in association with both a reduction in AMPD activity and an increase in AMPK activity. In summary, AMPD and AMPK are both important in hepatic fat accumulation and counter-regulate each other. We present the novel finding that uric acid inhibits AMPK kinase activity in fructose-fed hepatocytes thus providing new insights into the pathogenesis of fatty liver.
Conflict of interest statement
Figures
References
-
- Ford ES, Giles WH, Mokdad AH (2004) Increasing prevalence of the metabolic syndrome among u.s. Adults. Diabetes Care 27: 2444–2449. - PubMed
-
- Polyzos SA, Kountouras J, Deretzi G, Zavos C, Mantzoros CS (2012) The Emerging Role of Endocrine Disruptors in Pathogenesis of Insulin Resistance: A Concept Implicating Nonalcoholic Fatty Liver Disease. Curr Mol Med. - PubMed
-
- Musso G, Gambino R, Cassader M (2010) Emerging molecular targets for the treatment of nonalcoholic fatty liver disease. Annu Rev Med 61: 375–392. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
