

Inflammation and diabetes-accelerated atherosclerosis: myeloid cell mediators
- PMID: 23153419
- PMCID: PMC3578033
- DOI: 10.1016/j.tem.2012.10.002
Inflammation and diabetes-accelerated atherosclerosis: myeloid cell mediators
Abstract
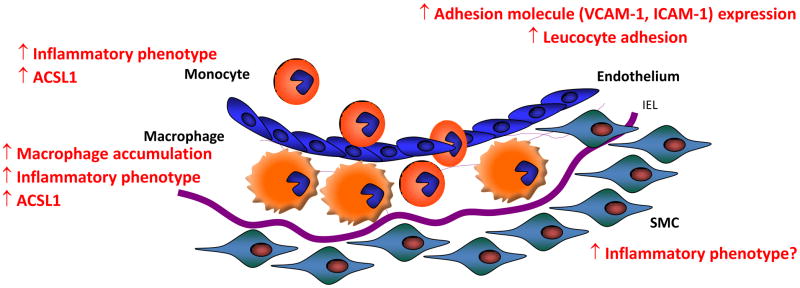
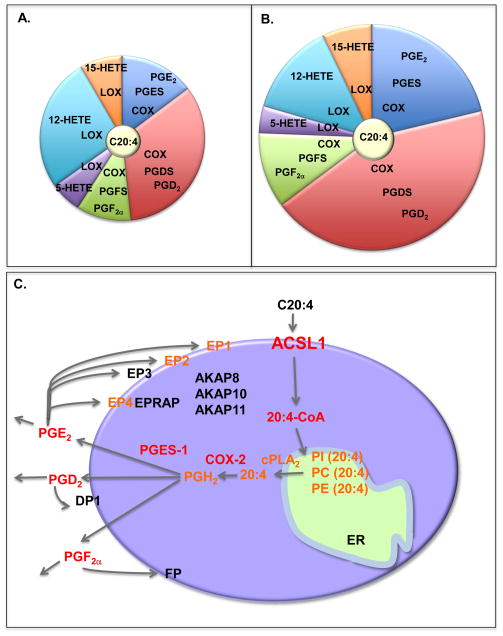
Monocytes and macrophages respond to and govern inflammation by producing a plethora of inflammatory modulators, including cytokines, chemokines, and arachidonic acid (C20:4)-derived lipid mediators. One of the most prevalent inflammatory diseases is cardiovascular disease, caused by atherosclerosis, and accelerated by diabetes. Recent research has demonstrated that monocytes/macrophages from diabetic mice and humans with type 1 diabetes show upregulation of the enzyme, acyl-CoA synthetase 1 (ACSL1), which promotes C20:4 metabolism, and that ACSL1 inhibition selectively protects these cells from the inflammatory and proatherosclerotic effects of diabetes, in mice. Increased understanding of the role of ACSL1 and other culprits in monocytes/macrophages in inflammation and diabetes-accelerated atherosclerosis offers hope for new treatment strategies to combat diabetic vascular disease.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
