

Chromothripsis: chromosomes in crisis
- PMID: 23153487
- PMCID: PMC3514072
- DOI: 10.1016/j.devcel.2012.10.010
Chromothripsis: chromosomes in crisis
Abstract
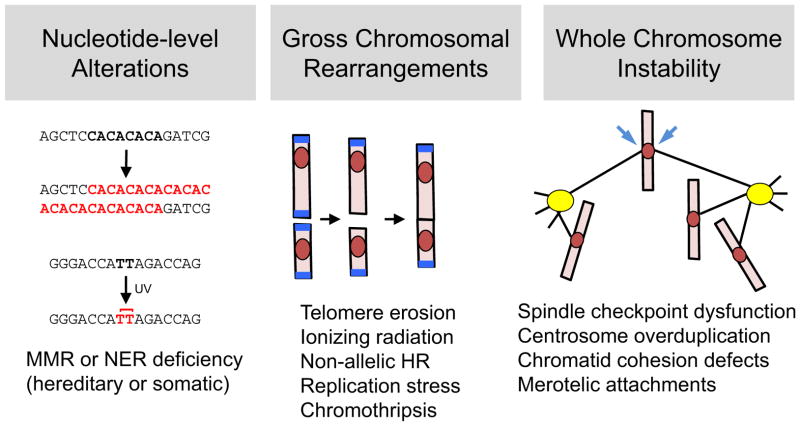
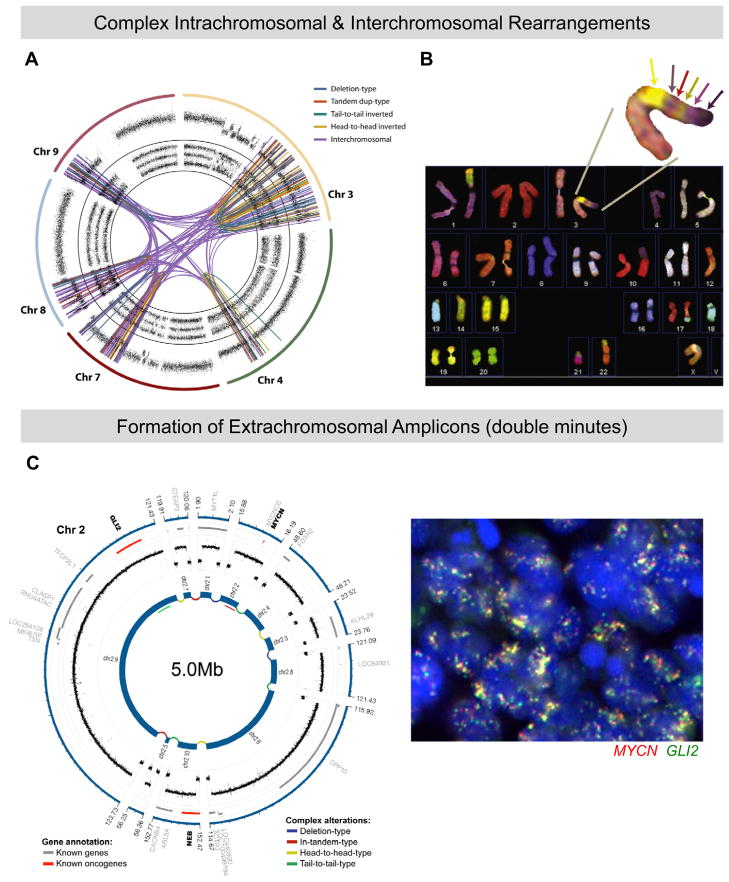
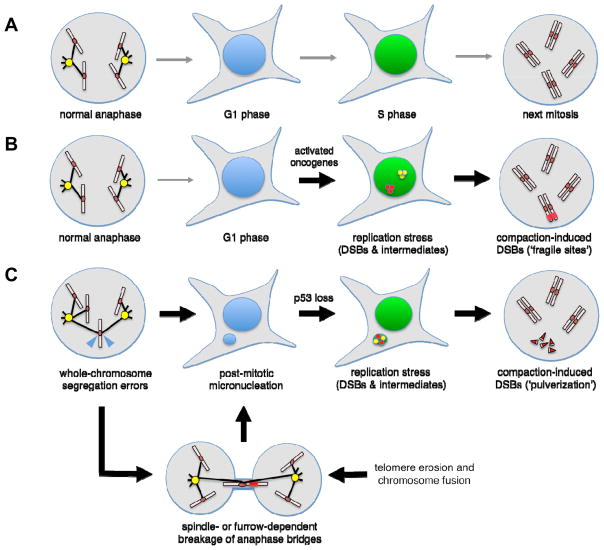
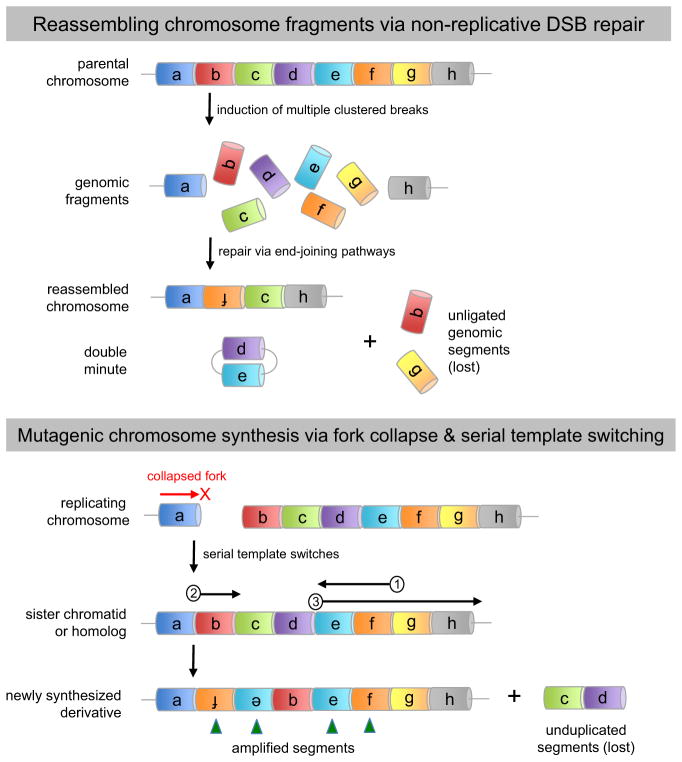
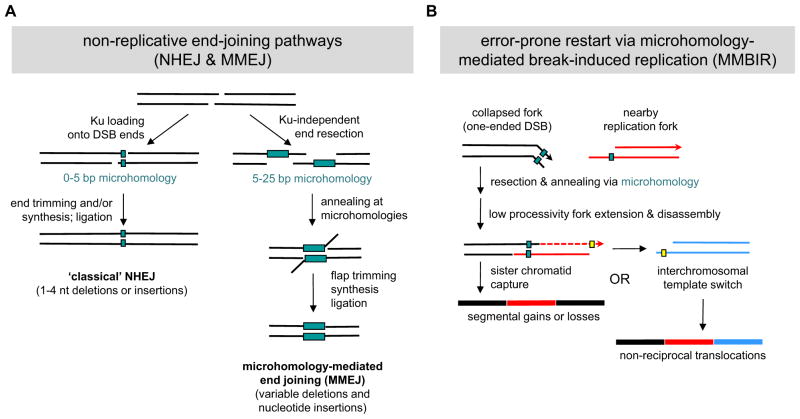
During oncogenesis, cells acquire multiple genetic alterations that confer essential tumor-specific traits, including immortalization, escape from antimitogenic signaling, neovascularization, invasiveness, and metastatic potential. In most instances, these alterations are thought to arise incrementally over years, if not decades. However, recent progress in sequencing cancer genomes has begun to challenge this paradigm, because a radically different phenomenon, termed chromothripsis, has been suggested to cause complex intra- and interchromosomal rearrangements on short timescales. In this Review, we review established pathways crucial for genome integrity and discuss how their dysfunction could precipitate widespread chromosome breakage and rearrangement in the course of malignancy.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. - PubMed
-
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nature reviews Molecular cell biology. 2004;5:792–804. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
