

Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas
- PMID: 23153539
- PMCID: PMC3713778
- DOI: 10.1016/j.ccr.2012.10.009
Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas
Abstract
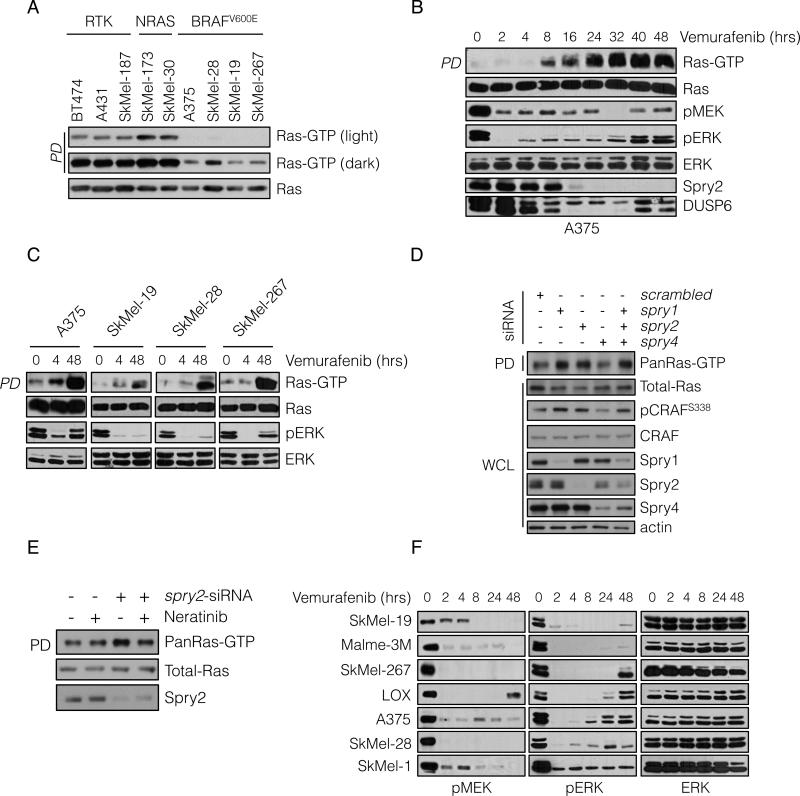
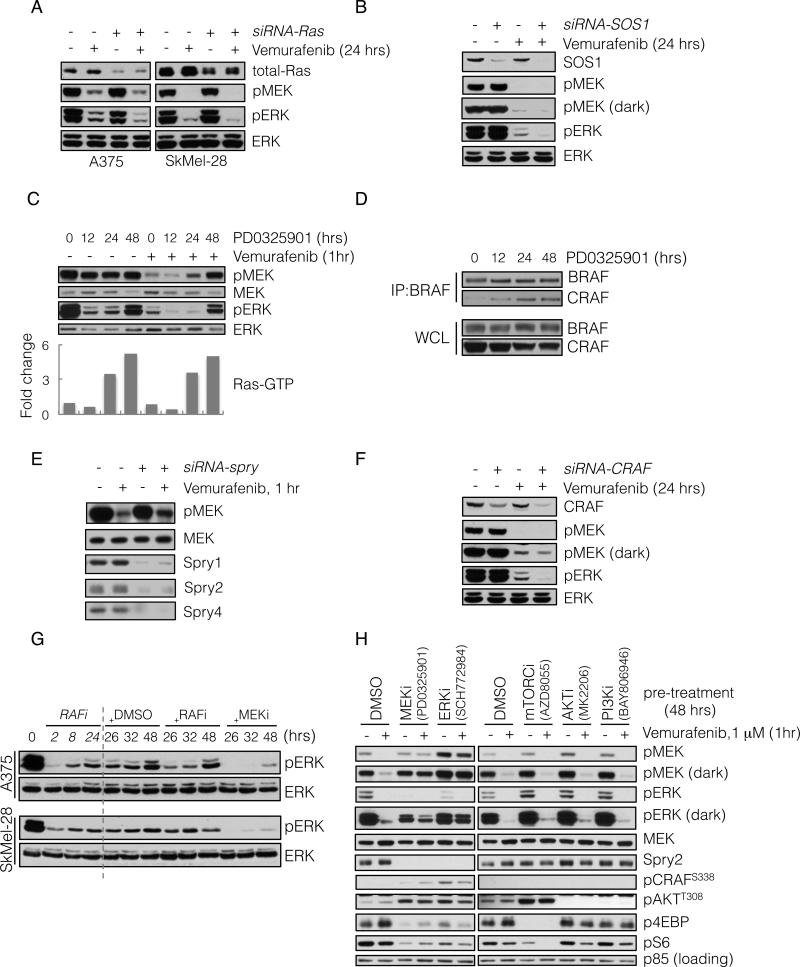
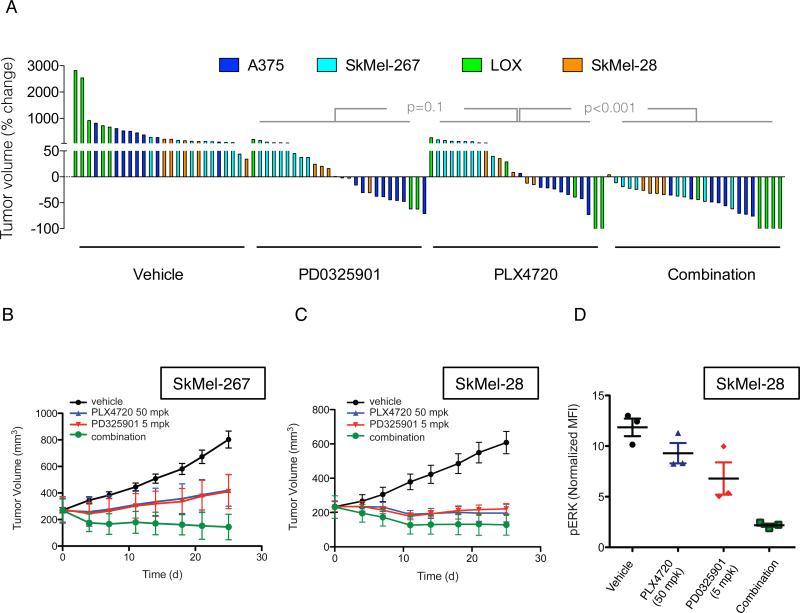
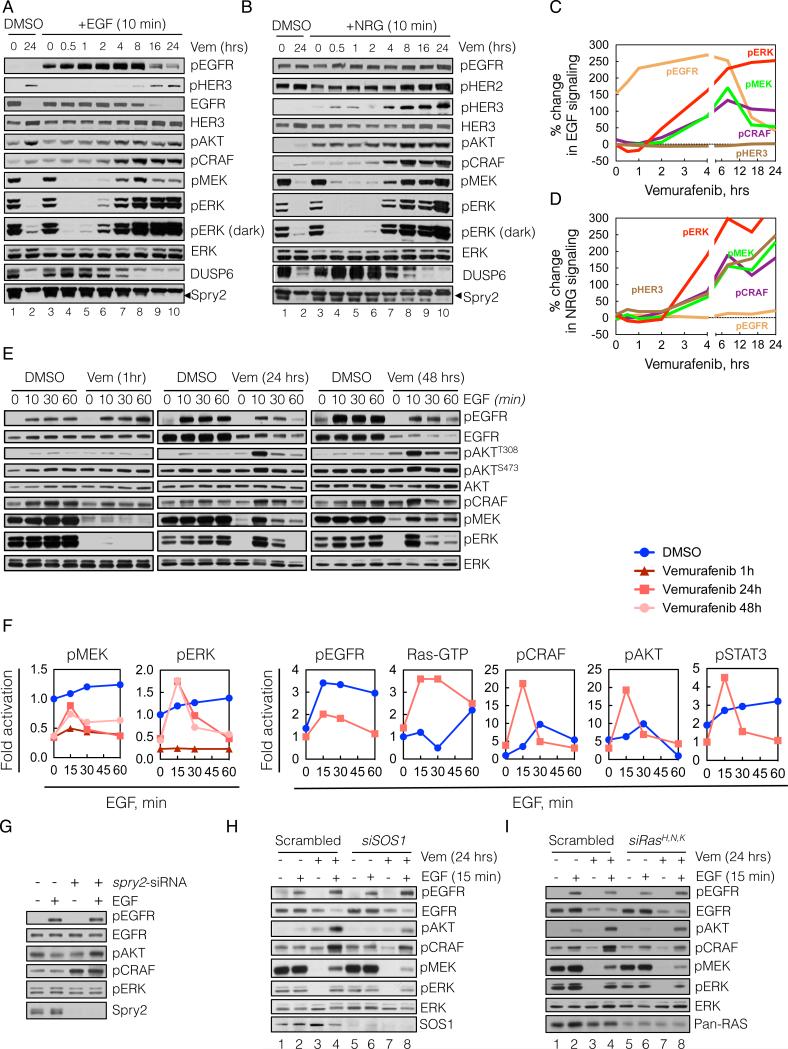
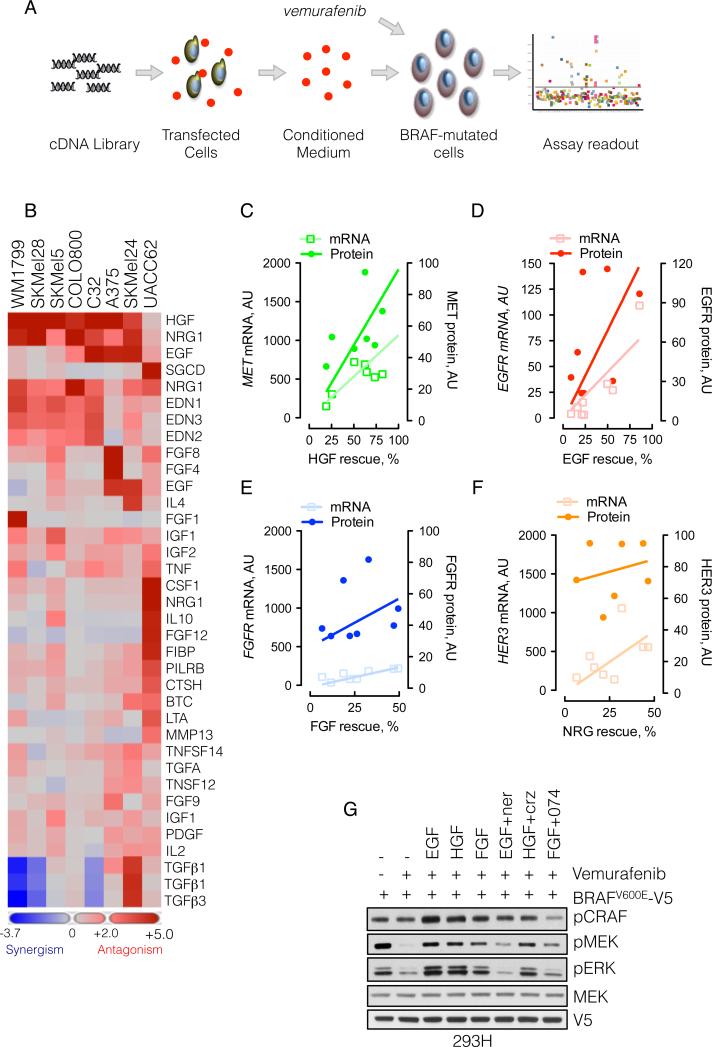
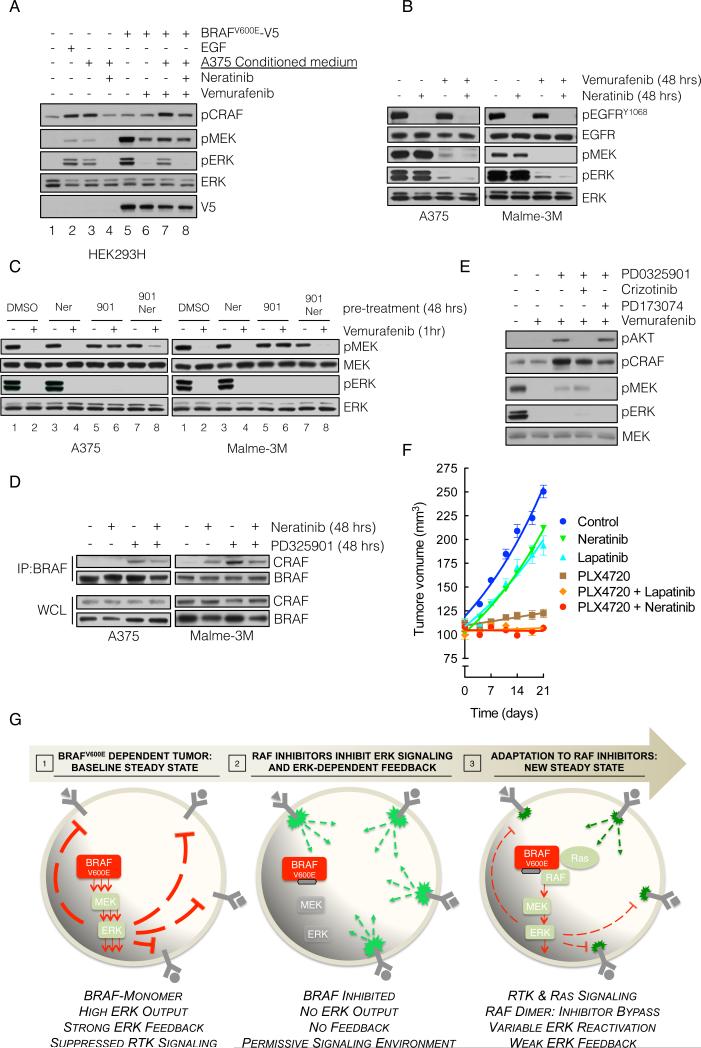
BRAF(V600E) drives tumors by dysregulating ERK signaling. In these tumors, we show that high levels of ERK-dependent negative feedback potently suppress ligand-dependent mitogenic signaling and Ras function. BRAF(V600E) activation is Ras independent and it signals as a RAF-inhibitor-sensitive monomer. RAF inhibitors potently inhibit RAF monomers and ERK signaling, causing relief of ERK-dependent feedback, reactivation of ligand-dependent signal transduction, increased Ras-GTP, and generation of RAF-inhibitor-resistant RAF dimers. This results in a rebound in ERK activity and culminates in a new steady state, wherein ERK signaling is elevated compared to its initial nadir after RAF inhibition. In this state, ERK signaling is RAF inhibitor resistant, and MEK inhibitor sensitive, and combined inhibition results in enhancement of ERK pathway inhibition and antitumor activity.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nature reviews Molecular cell biology. 2011;12:104–117. - PubMed
-
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–39866. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
