

Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies
- PMID: 23155049
- PMCID: PMC3543013
- DOI: 10.1074/jbc.M112.409250
Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies
Abstract
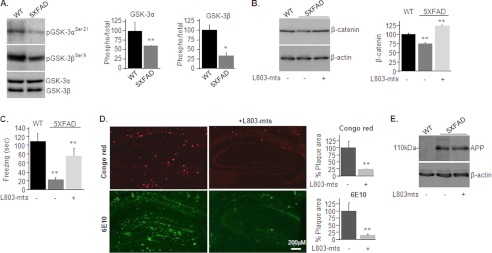
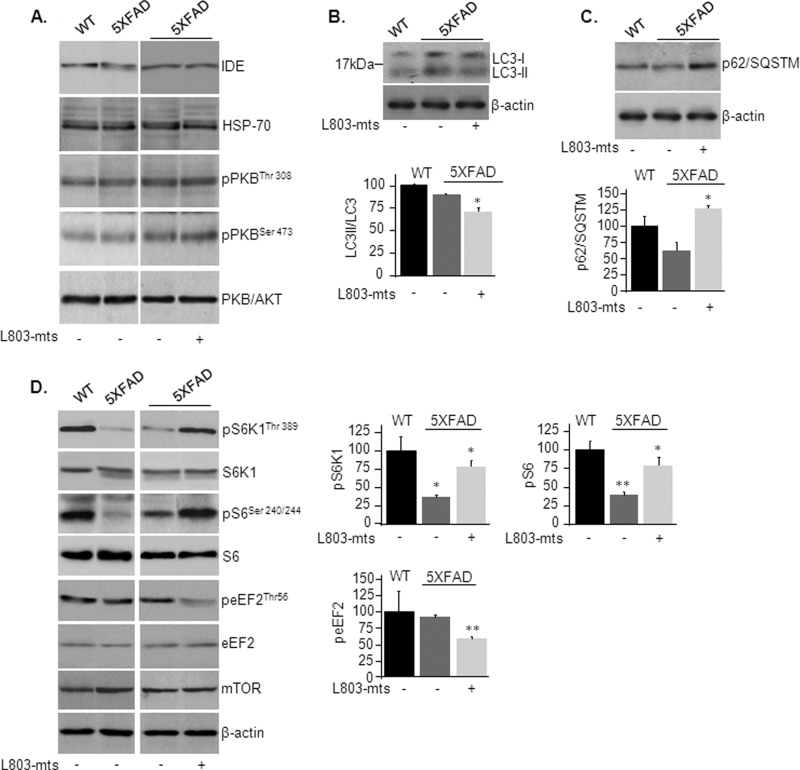
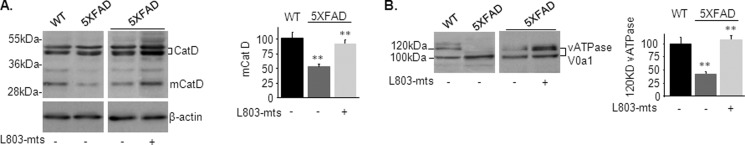
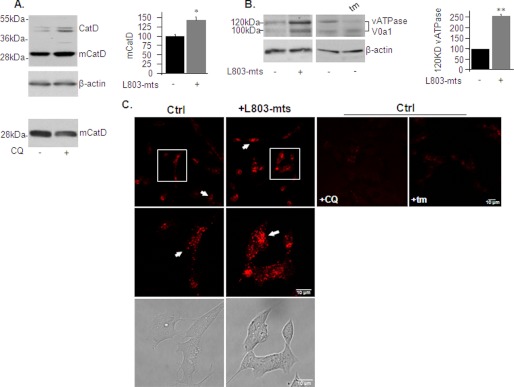
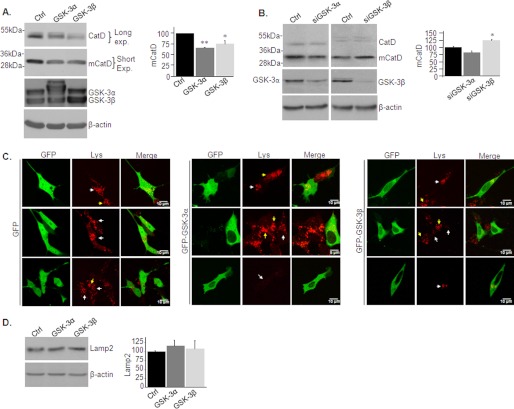
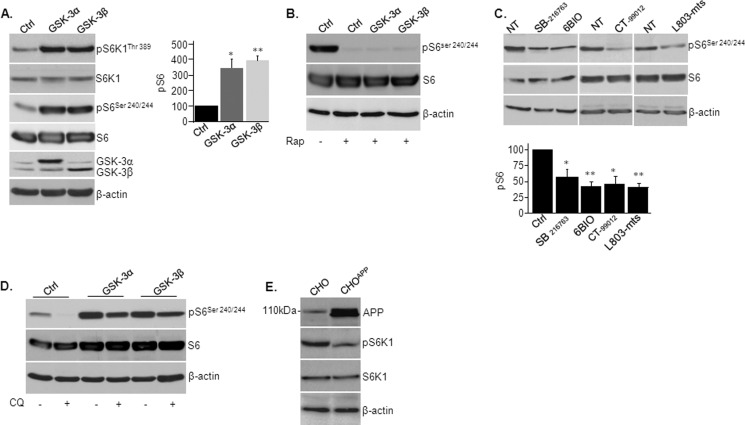
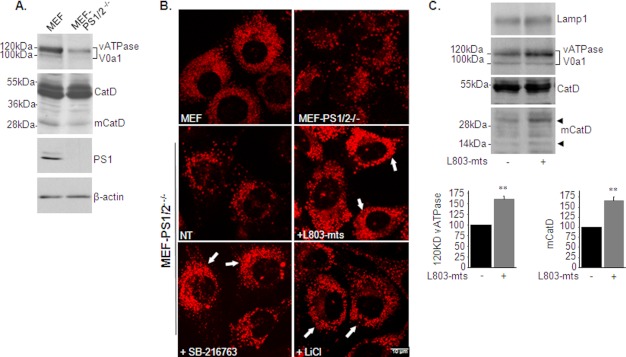
Accumulation of β-amyloid (Aβ) deposits is a primary pathological feature of Alzheimer disease that is correlated with neurotoxicity and cognitive decline. The role of glycogen synthase kinase-3 (GSK-3) in Alzheimer disease pathogenesis has been debated. To study the role of GSK-3 in Aβ pathology, we used 5XFAD mice co-expressing mutated amyloid precursor protein and presenilin-1 that develop massive cerebral Aβ loads. Both GSK-3 isozymes (α/β) were hyperactive in this model. Nasal treatment of 5XFAD mice with a novel substrate competitive GSK-3 inhibitor, L803-mts, reduced Aβ deposits and ameliorated cognitive deficits. Analyses of 5XFAD hemi-brain samples indicated that L803-mts restored the activity of mammalian target of rapamycin (mTOR) and inhibited autophagy. Lysosomal acidification was impaired in the 5XFAD brains as indicated by reduced cathepsin D activity and decreased N-glycoyslation of the vacuolar ATPase subunit V0a1, a modification required for lysosomal acidification. Treatment with L803-mts restored lysosomal acidification in 5XFAD brains. Studies in SH-SY5Y cells confirmed that GSK-3α and GSK-3β impair lysosomal acidification and that treatment with L803-mts enhanced the acidic lysosomal pool as demonstrated in LysoTracker Red-stained cells. Furthermore, L803-mts restored impaired lysosomal acidification caused by dysfunctional presenilin-1. We provide evidence that mTOR is a target activated by GSK-3 but inhibited by impaired lysosomal acidification and elevation in amyloid precursor protein/Aβ loads. Taken together, our data indicate that GSK-3 is a player in Aβ pathology. Inhibition of GSK-3 restores lysosomal acidification that in turn enables clearance of Aβ burdens and reactivation of mTOR. These changes facilitate amelioration in cognitive function.
Figures

References
-
- Selkoe D. J. (2001) Alzheimer's disease. Genes, proteins, and therapy. Physiol. Rev. 81, 741–766 - PubMed
-
- De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease. A multifactorial view on the disease process. Physiol. Rev. 90, 465–494 - PubMed
-
- Hardy J. (2009) The amyloid hypothesis for Alzheimer's disease. A critical reappraisal. J. Neurochem. 110, 1129–1134 - PubMed
-
- Näslund J., Haroutunian V., Mohs R., Davis K. L., Davies P., Greengard P., Buxbaum J. D. (2000) Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 283, 1571–1577 - PubMed
-
- Karran E., Mercken M., De Strooper B. (2011) The amyloid cascade hypothesis for Alzheimer's disease. An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
