

Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study
- PMID: 23158499
- PMCID: PMC3556524
- DOI: 10.1016/S1473-3099(12)70277-3
Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study
Abstract
Background: Tuberculosis incidence in the UK has risen in the past decade. Disease control depends on epidemiological data, which can be difficult to obtain. Whole-genome sequencing can detect microevolution within Mycobacterium tuberculosis strains. We aimed to estimate the genetic diversity of related M tuberculosis strains in the UK Midlands and to investigate how this measurement might be used to investigate community outbreaks.
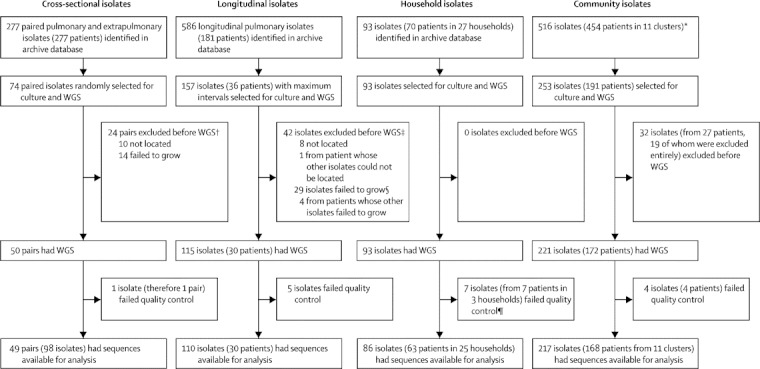
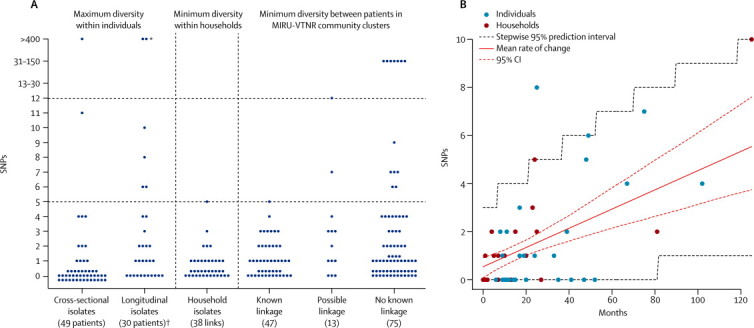
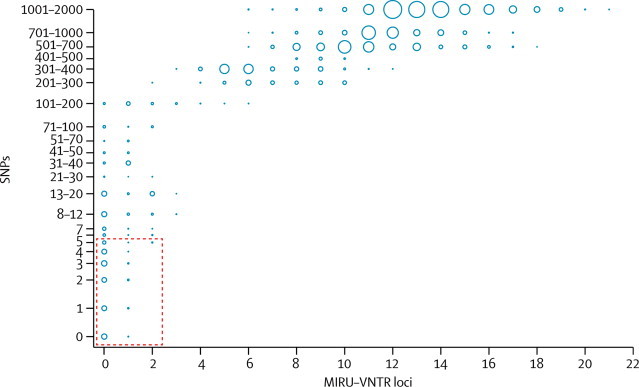
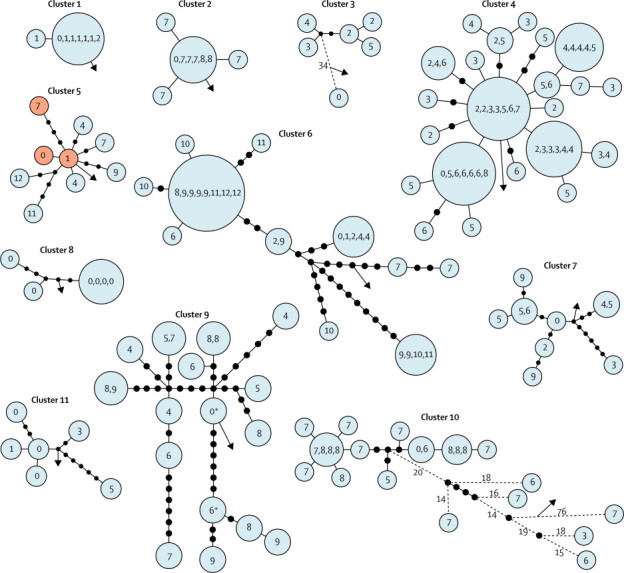
Methods: In a retrospective observational study, we used Illumina technology to sequence M tuberculosis genomes from an archive of frozen cultures. We characterised isolates into four groups: cross-sectional, longitudinal, household, and community. We measured pairwise nucleotide differences within hosts and between hosts in household outbreaks and estimated the rate of change in DNA sequences. We used the findings to interpret network diagrams constructed from 11 community clusters derived from mycobacterial interspersed repetitive-unit-variable-number tandem-repeat data.
Findings: We sequenced 390 separate isolates from 254 patients, including representatives from all five major lineages of M tuberculosis. The estimated rate of change in DNA sequences was 0.5 single nucleotide polymorphisms (SNPs) per genome per year (95% CI 0.3-0.7) in longitudinal isolates from 30 individuals and 25 families. Divergence is rarely higher than five SNPs in 3 years. 109 (96%) of 114 paired isolates from individuals and households differed by five or fewer SNPs. More than five SNPs separated isolates from none of 69 epidemiologically linked patients, two (15%) of 13 possibly linked patients, and 13 (17%) of 75 epidemiologically unlinked patients (three-way comparison exact p<0.0001). Genetic trees and clinical and epidemiological data suggest that super-spreaders were present in two community clusters.
Interpretation: Whole-genome sequencing can delineate outbreaks of tuberculosis and allows inference about direction of transmission between cases. The technique could identify super-spreaders and predict the existence of undiagnosed cases, potentially leading to early treatment of infectious patients and their contacts.
Funding: Medical Research Council, Wellcome Trust, National Institute for Health Research, and the Health Protection Agency.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures

Comment in
-
Investigation of disease outbreaks with genome sequencing.Lancet Infect Dis. 2013 Feb;13(2):101-2. doi: 10.1016/S1473-3099(12)70295-5. Epub 2012 Nov 15. Lancet Infect Dis. 2013. PMID: 23158498 No abstract available.
-
An epidemiological view of microbial genomic data.Lancet Infect Dis. 2013 Feb;13(2):104-5. doi: 10.1016/S1473-3099(12)70324-9. Lancet Infect Dis. 2013. PMID: 23347622 No abstract available.
References
-
- Health Protection Agency Tuberculosis in the UK: 2012 report. July 5, 2012. http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317134913404 (accessed Oct 9, 2012).
-
- Abubakar I, Lipman M, Anderson C, Davies P, Zumla A. Tuberculosis in the UK—time to regain control. BMJ. 2011;343:d4281. - PubMed
-
- Hawkey PM, Smith EG, Evans JT. Mycobacterial interspersed repetitive unit typing of Mycobacterium tuberculosis compared to IS6110-based restriction fragment length polymorphism analysis for investigation of apparently clustered cases of tuberculosis. J Clin Microbiol. 2003;41:3514–3520. - PMC - PubMed
-
- Li J, Driver CR, Munsiff SS, Fujiwara PI. Finding contacts of homeless tuberculosis patients in New York City. Int J Tuberc Lung Dis. 2003;7:S397–S404. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
