

doi: 10.1038/nmeth.2251.
Epub 2012 Nov 18.
Streaming fragment assignment for real-time analysis of sequencing experiments
Affiliations
- PMID: 23160280
- PMCID: PMC3880119
- DOI: 10.1038/nmeth.2251
Item in Clipboard
Streaming fragment assignment for real-time analysis of sequencing experiments
Nat Methods.
2013 Jan.
Abstract
We present eXpress, a software package for efficient probabilistic assignment of ambiguously mapping sequenced fragments. eXpress uses a streaming algorithm with linear run time and constant memory use. It can determine abundances of sequenced molecules in real time and can be applied to ChIP-seq, metagenomics and other large-scale sequencing data. We demonstrate its use on RNA-seq data and show that eXpress achieves greater efficiency than other quantification methods.
Figures
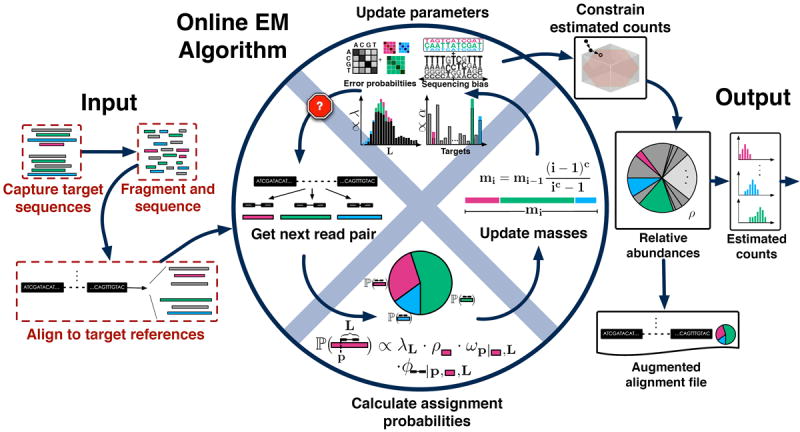
Overview of eXpress. The input consists of either single or paired-end reads aligned to a set of target sequences and provided in a file or streamed to eXpress. For single fragments that map to multiple sites, assignment probabilities are calculated for each site given previous estimates of target sequence abundances (initially a uniform prior is used). Next, a “forgetting mass” is calculated and partial counts are distributed to the target sequences according to the assignment probability. Parameters for fragment length distribution, sequence bias, and sequence read errors are updated in a similar fashion and used in the next round of alignment. Once the input data has been processed, relative abundances are calculated from the count distributions, along with distributions of estimated and effective counts. An alignment file that includes mapping probabilities can be generated. eXpress can determine whether further sequencing is needed by monitoring relative abundances, making it applicable to real-time sequencing and analysis.
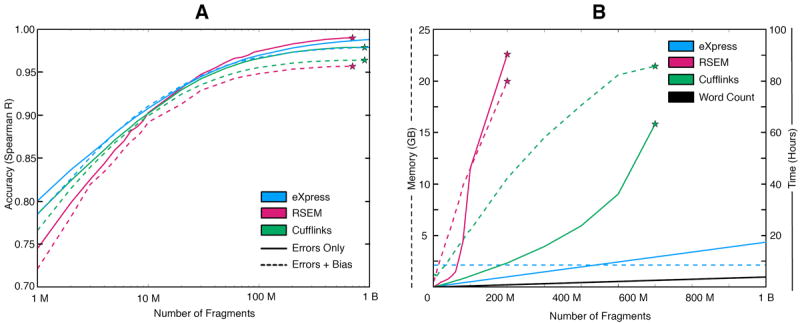
(a) Accuracy of eXpress, RSEM, and Cufflinks at multiple sequencing depths in a simulation of one billion read pair fragments generated with (dashed lines) and without (solid lines) sequencing bias. Accuracy for different abundance levels can be found in Supplementary Figure 4. (b) Comparison of time and memory requirements. Since eXpress only stores counts for each of the targets and auxiliary parameters, its memory use is constant in the number of fragments processed. The running time scales linearly with the number of fragments. Stars represent an imposed memory constraint of 24 GB or a software crash
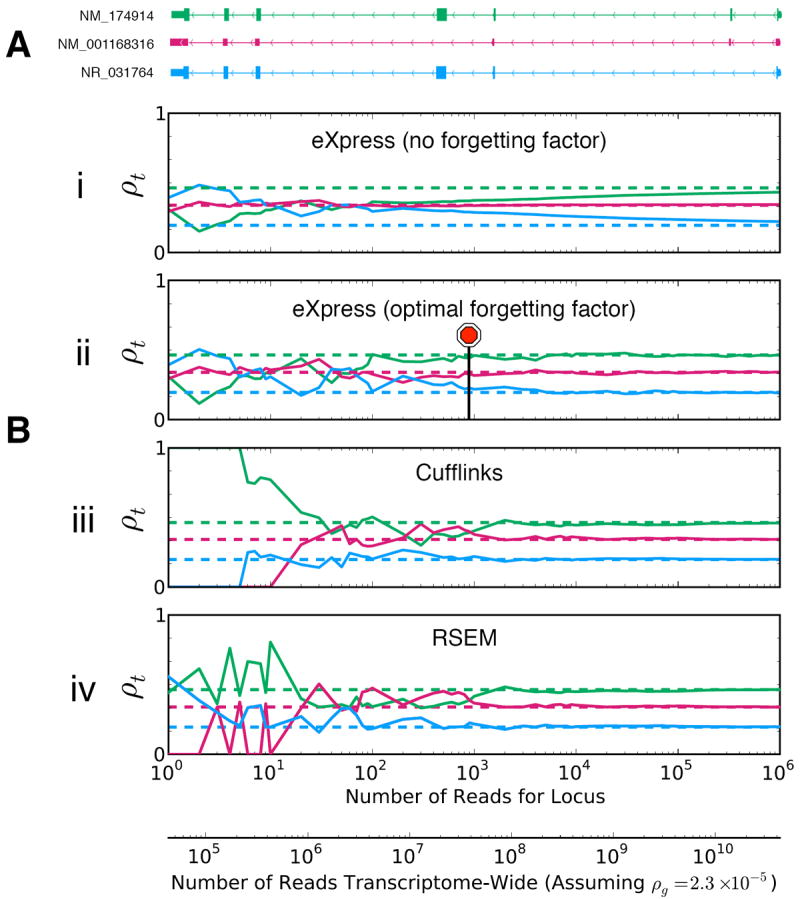
Example of abundance estimation by eXpress, RSEM, and Cufflinks at different depths of simulated data for the three-isoform human gene UGT3A2. The RefSeq annotation is shown at top. Dashed lines indicate the ground-truth relative abundances used for the simulation. eXpress only processes each fragment once whereas RSEM and Cufflinks perform many iterations before converging to the maximum likelihood solution. Nevertheless, as more fragments are observed, all three algorithms converge toward the correct answer at approximately the same depth. In fact, eXpress is more robust than the batch algorithms at low depth due to its use of a prior. The stop sign shows where eXpress using an optimal forgetting factor would automatically stop if a convergence threshold was set to 10-6 in terms of the Kullback-Leibler divergence between the abundance estimates at intervals of 100 fragments. The lower x-axis shows the estimated depth required to observe the corresponding number of reads mapping to this gene (upper x-axis) at a fixed gene-level abundance. Abundance was calculated using a human embryonic stem cell RNA-seq dataset (Online Methods).
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
