

Identifying reference genes with stable expression from high throughput sequence data
- PMID: 23162540
- PMCID: PMC3494082
- DOI: 10.3389/fmicb.2012.00385
Identifying reference genes with stable expression from high throughput sequence data
Abstract
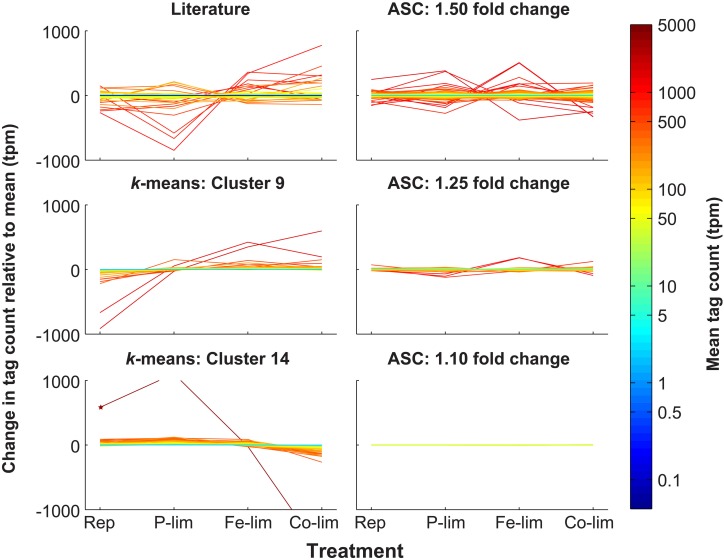
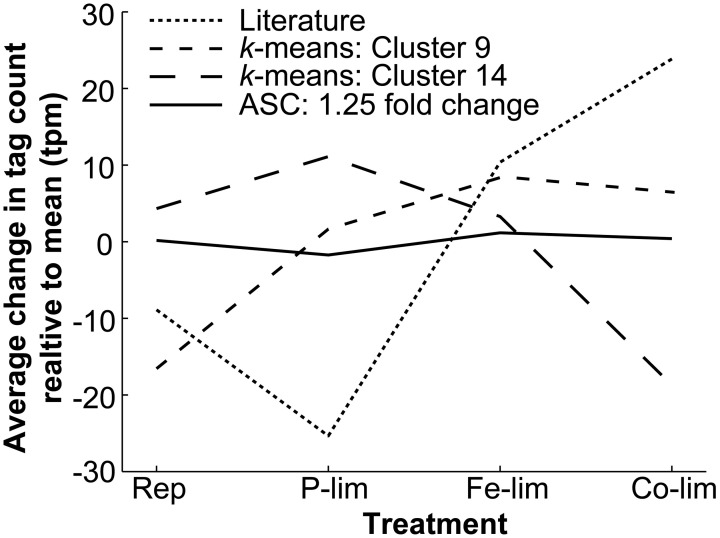
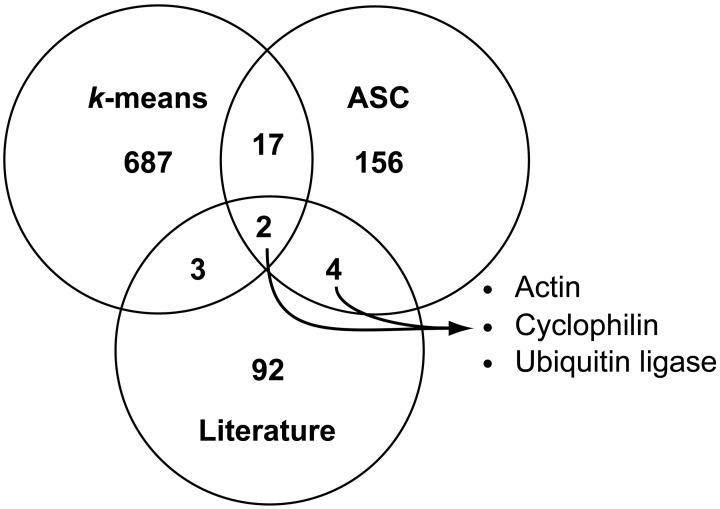
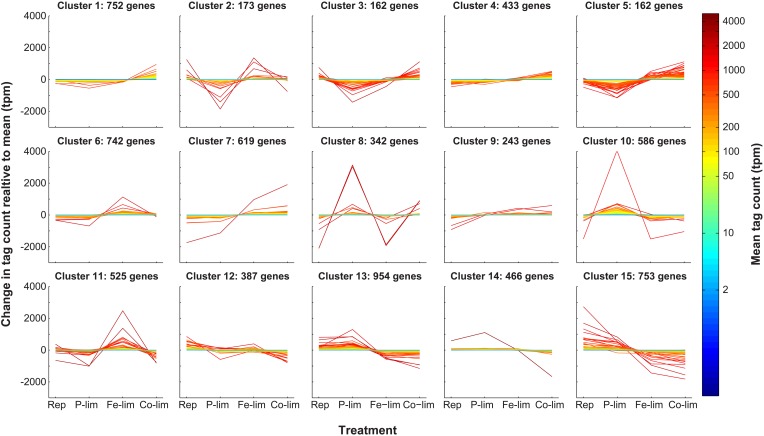
Genes that are constitutively expressed across multiple environmental stimuli are crucial to quantifying differentially expressed genes, particularly when employing quantitative reverse transcriptase polymerase chain reaction (RT-qPCR) assays. However, the identification of these potential reference genes in non-model organisms is challenging and is often guided by expression patterns in distantly related organisms. Here, transcriptome datasets from the diatom Thalassiosira pseudonana grown under replete, phosphorus-limited, iron-limited, and phosphorus and iron co-limited nutrient regimes were analyzed through literature-based searches for homologous reference genes, k-means clustering, and analysis of sequence counts (ASC) to identify putative reference genes. A total of 9759 genes were identified and screened for stable expression. Literature-based searches surveyed 18 generally accepted reference genes, revealing 101 homologs in T. pseudonana with variable expression and a wide range of mean tags per million. k-means analysis parsed the whole transcriptome into 15 clusters. The two most stable clusters contained 709 genes, but still had distinct patterns in expression. ASC analyses identified 179 genes that were stably expressed (posterior probability < 0.1 for 1.25 fold change). Genes known to have a stable expression pattern across the test treatments, like actin, were identified in this pool of 179 candidate genes. ASC can be employed on data without biological replicates and was more robust than the k-means approach in isolating genes with stable expression. The intersection of the genes identified through ASC with commonly used reference genes from the literature suggests that actin and ubiquitin ligase may be useful reference genes for T. pseudonana and potentially other diatoms. With the wealth of transcriptome sequence data becoming available, ASC can be easily applied to transcriptome datasets from other phytoplankton to identify reference genes.
Keywords: RT-qPCR; Thalassiosira pseudonana; diatom; housekeeping genes; phytoplankton; reference gene; relative gene expression; transcriptome.
Figures
References
-
- Antonov J., Goldstein D. R., Oberli A., Baltzer A., Pirotta M., Fleischmann A., et al. (2005). Reliable gene expression measurements from degraded RNA by quantitative real-time PCR depend on short amplicons and a proper normalization. Lab. Invest. 85, 1040–1050 10.1038/labinvest.3700303 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous