

Age-by-disease biological interactions: implications for late-life depression
- PMID: 23162569
- PMCID: PMC3499806
- DOI: 10.3389/fgene.2012.00237
Age-by-disease biological interactions: implications for late-life depression
Abstract
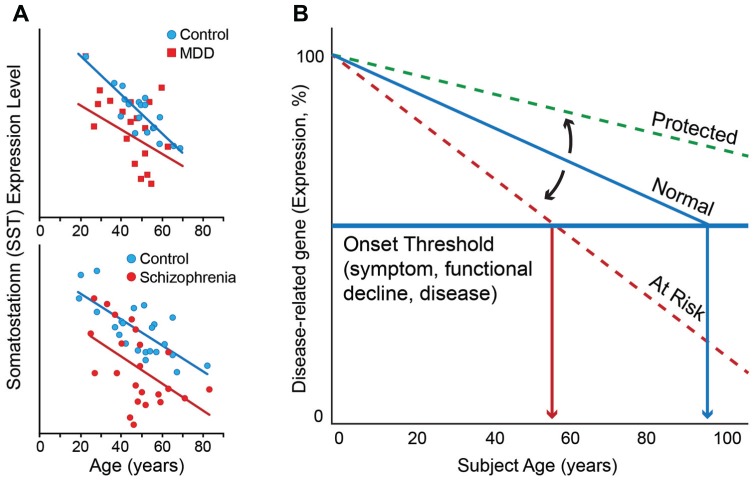
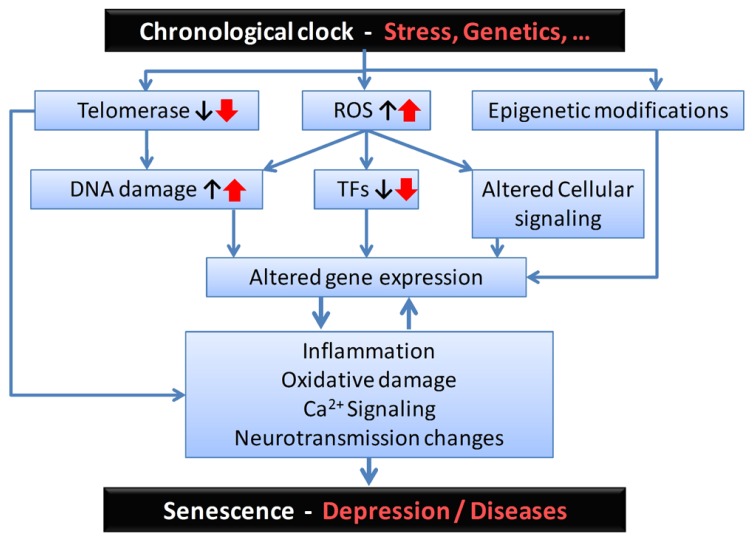
Onset of depressive symptoms after the age of 65, or late-life depression (LLD), is common and poses a significant burden on affected individuals, caretakers, and society. Evidence suggests a unique biological basis for LLD, but current hypotheses do not account for its pathophysiological complexity. Here we propose a novel etiological framework for LLD, the age-by-disease biological interaction hypothesis, based on the observations that the subset of genes that undergoes lifelong progressive changes in expression is restricted to a specific set of biological processes, and that a disproportionate number of these age-dependent genes have been previously and similarly implicated in neurodegenerative and neuropsychiatric disorders, including depression. The age-by-disease biological interaction hypothesis posits that age-dependent biological processes (i) are "pushed" in LLD-promoting directions by changes in gene expression naturally occurring during brain aging, which (ii) directly contribute to pathophysiological mechanisms of LLD, and (iii) that individual variability in rates of age-dependent changes determines risk or resiliency to develop age-related disorders, including LLD. We review observations supporting this hypothesis, including consistent and specific age-dependent changes in brain gene expression and their overlap with neuropsychiatric and neurodegenerative disease pathways. We then review preliminary reports supporting the genetic component of this hypothesis. Other potential biological mediators of age-dependent gene changes are proposed. We speculate that studies examining the relative contribution of these mechanisms to age-dependent changes and related disease mechanisms will not only provide critical information on the biology of normal aging of the human brain, but will inform our understanding of age-dependent diseases, in time fostering the development of new interventions for prevention and treatment of age-dependent diseases, including LLD.
Keywords: depression; epigenetic modifications; gene expression; late-life depression; molecular aging; oxidative stress; telomere.
Figures
Similar articles
-
The age-by-disease interaction hypothesis of late-life depression.Am J Geriatr Psychiatry. 2013 May;21(5):418-32. doi: 10.1016/j.jagp.2013.01.053. Epub 2013 Feb 6. Am J Geriatr Psychiatry. 2013. PMID: 23570886 Free PMC article. Review.
-
Molecular aging of the brain, neuroplasticity, and vulnerability to depression and other brain-related disorders.Dialogues Clin Neurosci. 2013 Mar;15(1):53-65. doi: 10.31887/DCNS.2013.15.1/esibille. Dialogues Clin Neurosci. 2013. PMID: 23576889 Free PMC article. Review.
-
Shared Genetic Risk Factors for Late-Life Depression and Alzheimer's Disease.J Alzheimers Dis. 2016 Mar 8;52(1):1-15. doi: 10.3233/JAD-151129. J Alzheimers Dis. 2016. PMID: 27060956 Review.
-
Depression in the elderly: brain correlates, neuropsychological findings, and role of vascular lesion load.Curr Opin Neurol. 2013 Dec;26(6):656-61. doi: 10.1097/WCO.0000000000000028. Curr Opin Neurol. 2013. PMID: 24184971 Review.
-
Plasma proteomic signature of major depressive episode in the elderly.J Proteomics. 2022 Oct 30;269:104713. doi: 10.1016/j.jprot.2022.104713. Epub 2022 Sep 1. J Proteomics. 2022. PMID: 36058540
Cited by
-
The Role of BDNF in Age-Dependent Changes of Excitatory and Inhibitory Synaptic Markers in the Human Prefrontal Cortex.Neuropsychopharmacology. 2016 Dec;41(13):3080-3091. doi: 10.1038/npp.2016.126. Epub 2016 Jul 15. Neuropsychopharmacology. 2016. PMID: 27417517 Free PMC article.
-
The role of the nervous system in aging and longevity.Front Genet. 2013 Jun 27;4:124. doi: 10.3389/fgene.2013.00124. Print 2013. Front Genet. 2013. PMID: 23818894 Free PMC article. No abstract available.
-
Hypermethylation of BDNF and SST Genes in the Orbital Frontal Cortex of Older Individuals: A Putative Mechanism for Declining Gene Expression with Age.Neuropsychopharmacology. 2015 Oct;40(11):2604-13. doi: 10.1038/npp.2015.107. Epub 2015 Apr 16. Neuropsychopharmacology. 2015. PMID: 25881116 Free PMC article.
-
Major depression and enhanced molecular senescence abnormalities in young and middle-aged adults.Transl Psychiatry. 2019 Aug 21;9(1):198. doi: 10.1038/s41398-019-0541-3. Transl Psychiatry. 2019. PMID: 31434875 Free PMC article.
-
Telomere shortening in late-life depression: A potential marker of depression severity.Brain Behav. 2021 Aug;11(8):e2255. doi: 10.1002/brb3.2255. Epub 2021 Jun 21. Brain Behav. 2021. PMID: 34152095 Free PMC article.
References
-
- Abe N., Uchida S., Otsuki K., Hobara T., Yamagata H., Higuchi F., et al. (2011). Altered sirtuin deacetylase gene expression in patients with a mood disorder. J. Psychiatr. Res. 45 1106–1112 - PubMed
-
- Ahmed S., Passos J. F., Birket M. J., Beckmann T., Brings S., Peters H., et al. (2008). Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell Sci. 121 1046–1053 - PubMed
-
- Alexopoulos G. S., Meyers B. S., Young R. C., Campbell S., Silbersweig D., Charlson M. (1997). ‘Vascular depression’ hypothesis. Arch. Gen. Psychiatry 54 915–922 - PubMed
-
- American Psychiatric Association. (2000). Diagnostic Criteria from DSM-IV-TR. Washington, DC: American Psychiatric Association
Grants and funding
LinkOut - more resources
Full Text Sources