

Comparative genomics and stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli
- PMID: 23162798
- PMCID: PMC3491183
- DOI: 10.3389/fcimb.2012.00133
Comparative genomics and stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli
Abstract
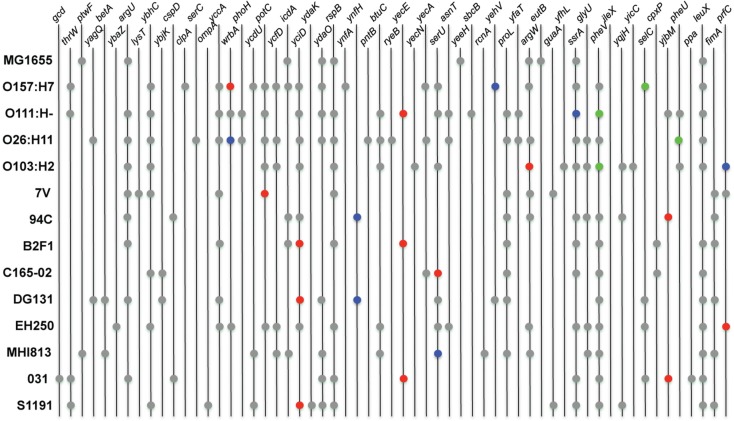
Infection by Escherichia coli and Shigella species are among the leading causes of death due to diarrheal disease in the world. Shiga toxin-producing E. coli (STEC) that do not encode the locus of enterocyte effacement (LEE-negative STEC) often possess Shiga toxin gene variants and have been isolated from humans and a variety of animal sources. In this study, we compare the genomes of nine LEE-negative STEC harboring various stx alleles with four complete reference LEE-positive STEC isolates. Compared to a representative collection of prototype E. coli and Shigella isolates representing each of the pathotypes, the whole genome phylogeny demonstrated that these isolates are diverse. Whole genome comparative analysis of the 13 genomes revealed that in addition to the absence of the LEE pathogenicity island, phage-encoded genes including non-LEE encoded effectors, were absent from all nine LEE-negative STEC genomes. Several plasmid-encoded virulence factors reportedly identified in LEE-negative STEC isolates were identified in only a subset of the nine LEE-negative isolates further confirming the diversity of this group. In combination with whole genome analysis, we characterized the lambdoid phages harboring the various stx alleles and determined their genomic insertion sites. Although the integrase gene sequence corresponded with genomic location, it was not correlated with stx variant, further highlighting the mosaic nature of these phages. The transcription of these phages in different genomic backgrounds was examined. Expression of the Shiga toxin genes, stx(1) and/or stx(2), as well as the Q genes, were examined with quantitative reverse transcriptase polymerase chain reaction assays. A wide range of basal and induced toxin induction was observed. Overall, this is a first significant foray into the genome space of this unexplored group of emerging and divergent pathogens.
Keywords: Escherichia coli; Shiga toxin; evolution; microbial genomics; phage.
Figures







References
-
- Asadulghani M., Ogura Y., Ooka T., Itoh T., Sawaguchi A., Iguchi A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5, e1000408.10.1371/journal.ppat.1000408 - DOI - PMC - PubMed
-
- Beutin L., Kruger U., Krause G., Miko A., Martin A., Strauch E. (2008). Evaluation of major types of Shiga toxin 2E-producing Escherichia coli bacteria present in food, pigs, and the environment as potential pathogens for humans. Appl. Environ. Microbiol. 74, 4806–481610.1128/AEM.00623-08 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources