

Partial adrenalectomy minimizes the need for long-term hormone replacement in pediatric patients with pheochromocytoma and von Hippel-Lindau syndrome
- PMID: 23164001
- PMCID: PMC3846393
- DOI: 10.1016/j.jpedsurg.2012.07.003
Partial adrenalectomy minimizes the need for long-term hormone replacement in pediatric patients with pheochromocytoma and von Hippel-Lindau syndrome
Abstract
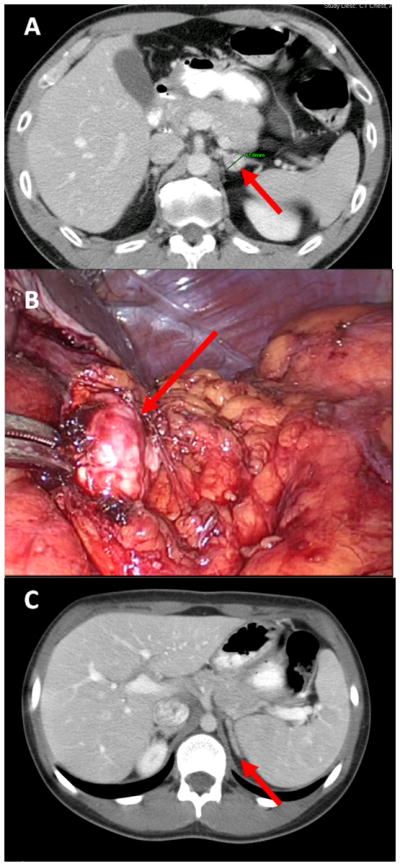
Purpose: Children with von Hippel-Lindau syndrome (VHL) are at an increased risk for developing bilateral pheochromocytomas. In an effort to illustrate the advantage of partial adrenalectomy (PA) over total adrenalectomy in children with VHL, we report the largest single series on PA for pediatric patients with VHL, demonstrating a balance between tumor removal and preservation of adrenocortical function.
Methods: From 1994 to 2011, a prospectively maintained database was reviewed to evaluate 10 pediatric patients with hereditary pheochromocytoma for PA. Surgery was performed if there was clinical evidence of pheochromocytoma and if normal adrenocortical tissue was evident on preoperative imaging and/or intraoperative ultrasonography. Perioperative data were collected, and patients were observed for postoperative steroid use and tumor recurrence.
Results: Ten pediatric patients with a diagnosis of VHL underwent 18 successful partial adrenalectomies (4 open, 14 laparoscopic). The median tumor size removed was 2.6 cm (range, 1.2-6.5 cm). Over a median follow-up of 7.2 years (range, 2.6-15.8 years), additional tumors in the ipsilateral adrenal gland were found in 2 patients. One patient underwent completion adrenalectomy, and 1 underwent a salvage PA with resection of the ipsilateral lesion. One patient required short-term steroid replacement therapy. At last follow-up, 7 patients had no radiographic or laboratory evidence of pheochromocytoma.
Conclusion: At our institution, PA is the preferred form of management for pheochromocytoma in the (VHL) pediatric population. This surgical approach allows for removal of tumor while preserving adrenocortical function and minimizing the adverse effects of long-term steroid replacement on puberty and quality of life.
Copyright © 2012. Published by Elsevier Inc.
Figures
References
-
- Walther MM, Keiser HR, Choyke PL, Rayford W, Lyne JC, Linehan WM. Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol. 1999 Feb;161(2):395–398. - PubMed
-
- Neumann HP, Berger DP, Sigmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 1993 Nov 18;329(21):1531–1538. - PubMed
-
- Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997 May;29(5):1133–1139. - PubMed
-
- Neumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. J Clin Endocrinol Metab. 1999 Aug;84(8):2608–2610. - PubMed
-
- Telenius-Berg M, Ponder MA, Berg B, Ponder BA, Werner S. Quality of life after bilateral adrenalectomy in MEN 2. Henry Ford Hosp Med J. 1989;37(3–4):160–163. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
