

Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway
- PMID: 23164821
- PMCID: PMC3873038
- DOI: 10.1038/mp.2012.163
Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway
Abstract
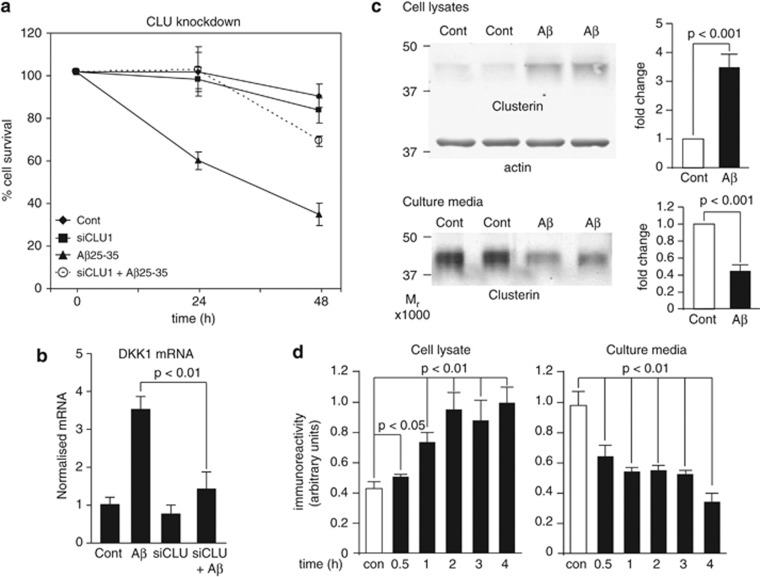
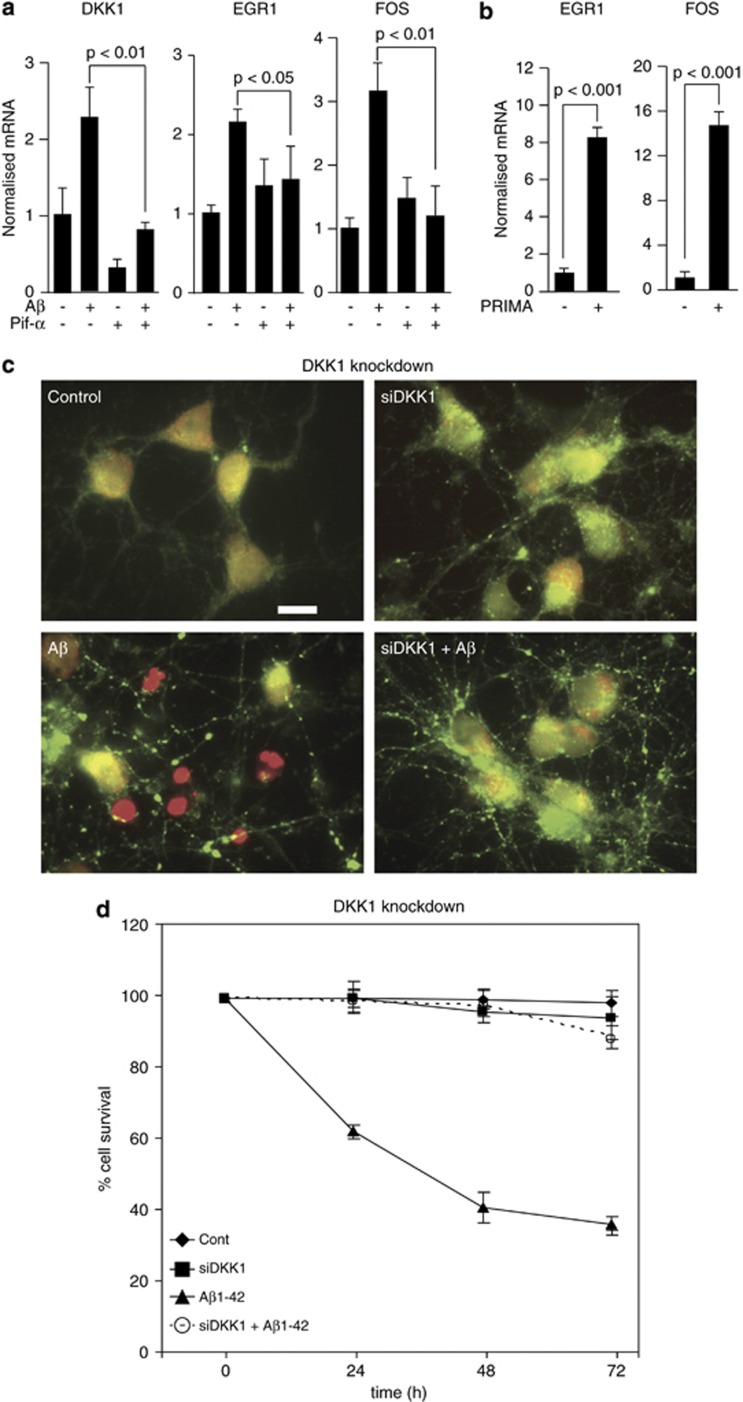
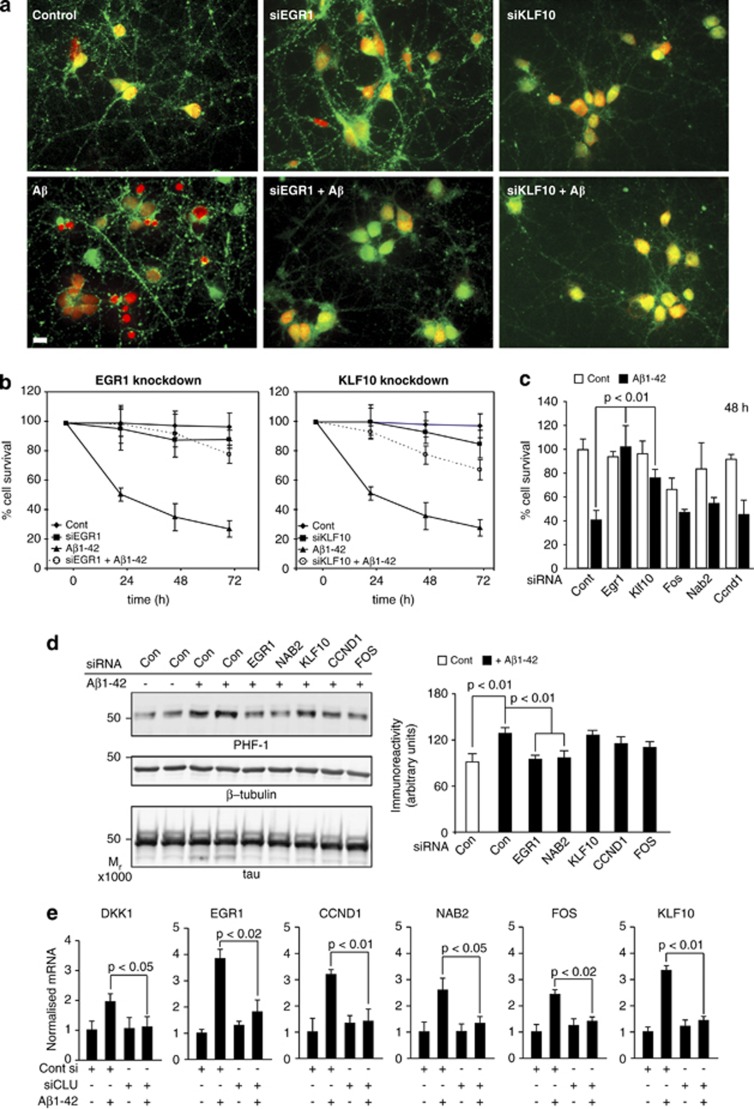
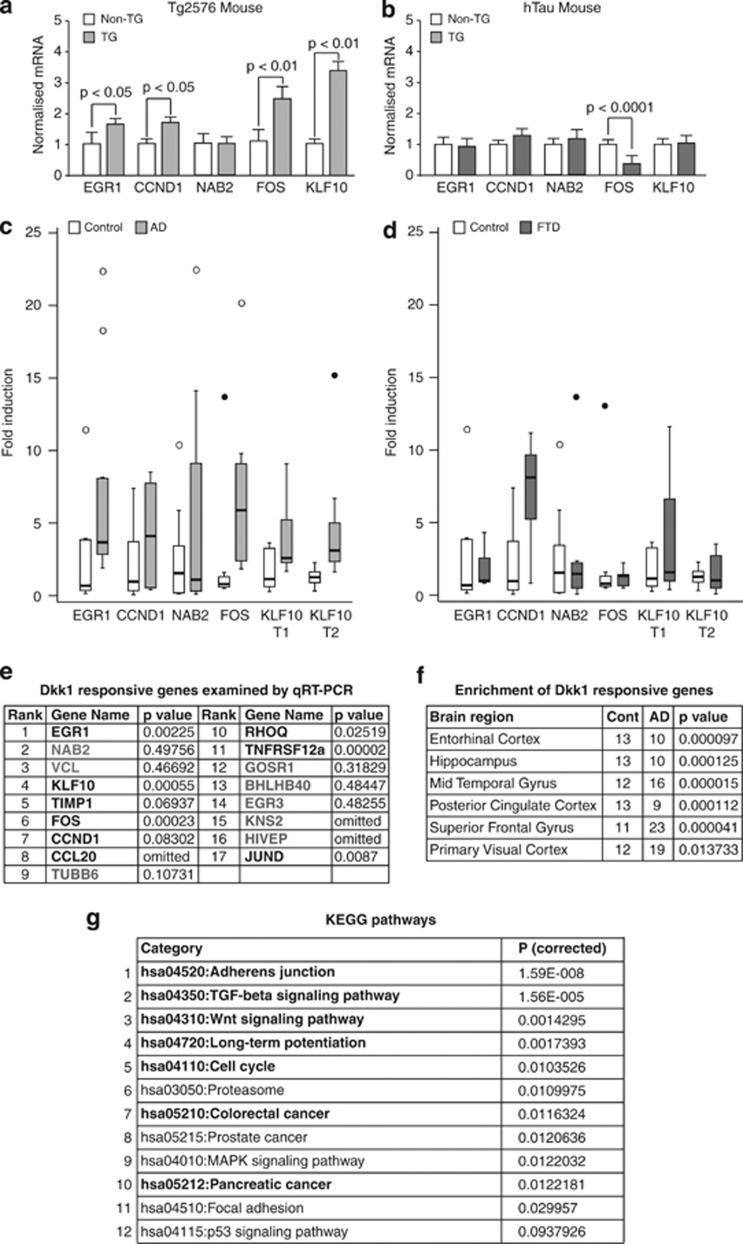
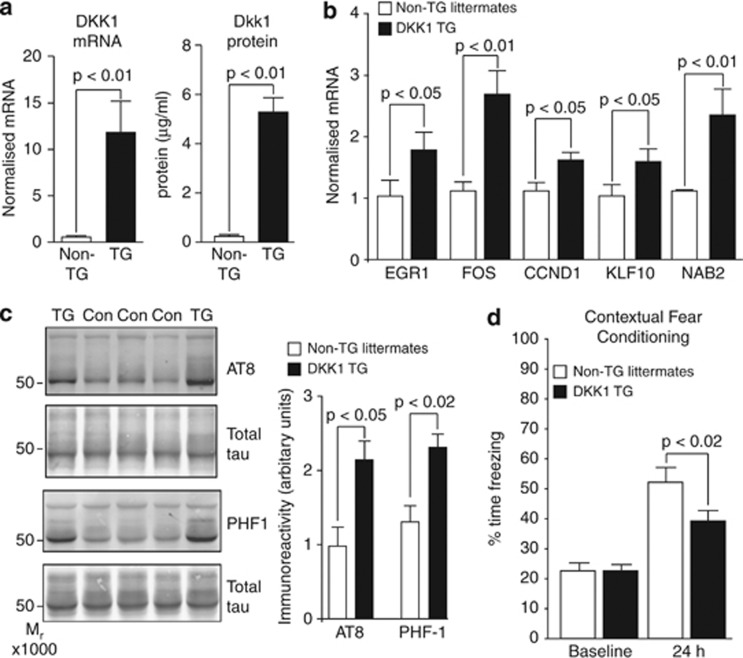
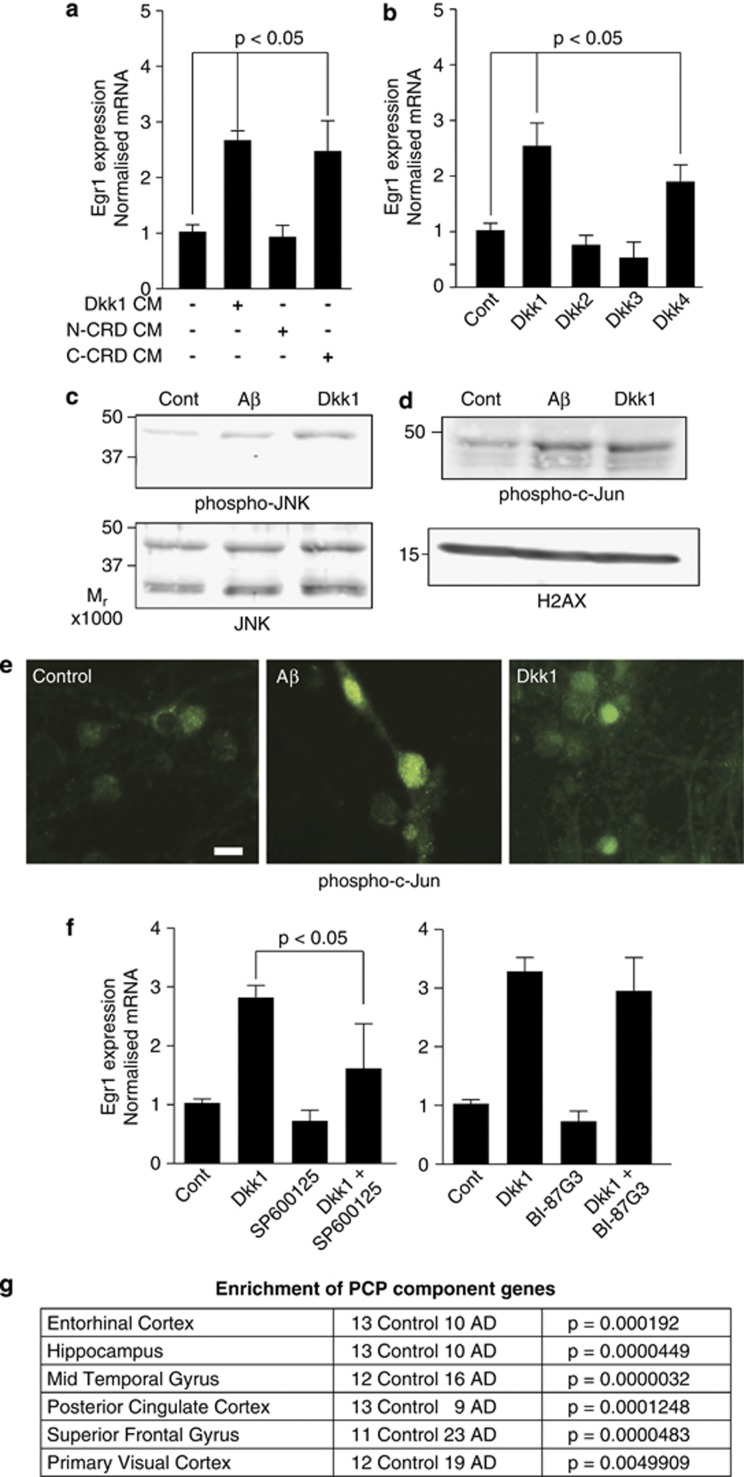
Although the mechanism of Aβ action in the pathogenesis of Alzheimer's disease (AD) has remained elusive, it is known to increase the expression of the antagonist of canonical wnt signalling, Dickkopf-1 (Dkk1), whereas the silencing of Dkk1 blocks Aβ neurotoxicity. We asked if clusterin, known to be regulated by wnt, is part of an Aβ/Dkk1 neurotoxic pathway. Knockdown of clusterin in primary neurons reduced Aβ toxicity and DKK1 upregulation and, conversely, Aβ increased intracellular clusterin and decreased clusterin protein secretion, resulting in the p53-dependent induction of DKK1. To further elucidate how the clusterin-dependent induction of Dkk1 by Aβ mediates neurotoxicity, we measured the effects of Aβ and Dkk1 protein on whole-genome expression in primary neurons, finding a common pathway suggestive of activation of wnt-planar cell polarity (PCP)-c-Jun N-terminal kinase (JNK) signalling leading to the induction of genes including EGR1 (early growth response-1), NAB2 (Ngfi-A-binding protein-2) and KLF10 (Krüppel-like factor-10) that, when individually silenced, protected against Aβ neurotoxicity and/or tau phosphorylation. Neuronal overexpression of Dkk1 in transgenic mice mimicked this Aβ-induced pathway and resulted in age-dependent increases in tau phosphorylation in hippocampus and cognitive impairment. Furthermore, we show that this Dkk1/wnt-PCP-JNK pathway is active in an Aβ-based mouse model of AD and in AD brain, but not in a tau-based mouse model or in frontotemporal dementia brain. Thus, we have identified a pathway whereby Aβ induces a clusterin/p53/Dkk1/wnt-PCP-JNK pathway, which drives the upregulation of several genes that mediate the development of AD-like neuropathologies, thereby providing new mechanistic insights into the action of Aβ in neurodegenerative diseases.
Figures
References
-
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. - PubMed
-
- Brion JP, Flament-Durand J, Dustin P. Alzheimer's disease and tau proteins. Lancet. 1986;2:1098. - PubMed
-
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. - PubMed
-
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
