

TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis
- PMID: 23166301
- PMCID: PMC3554219
- DOI: 10.1128/MCB.01225-12
TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis
Abstract
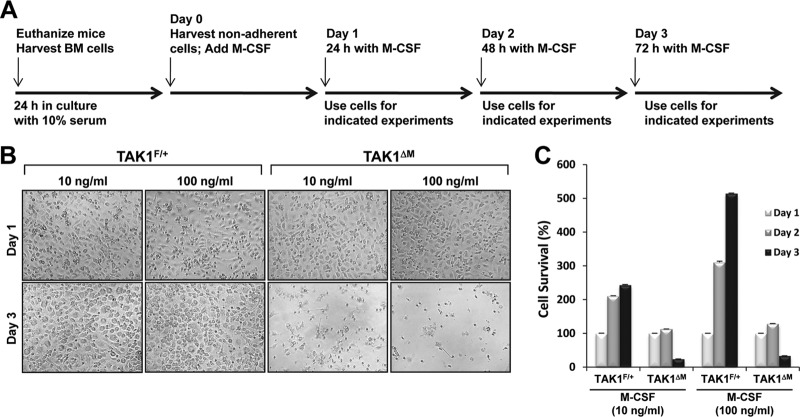
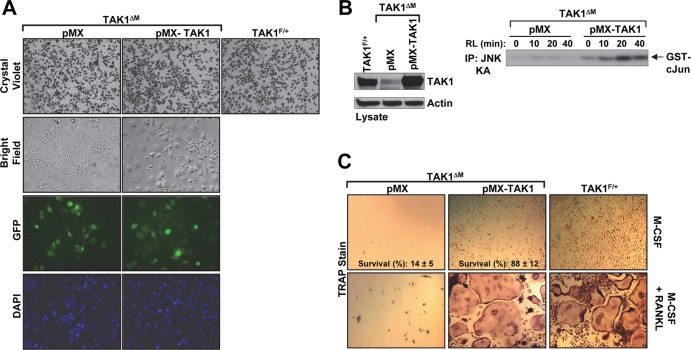
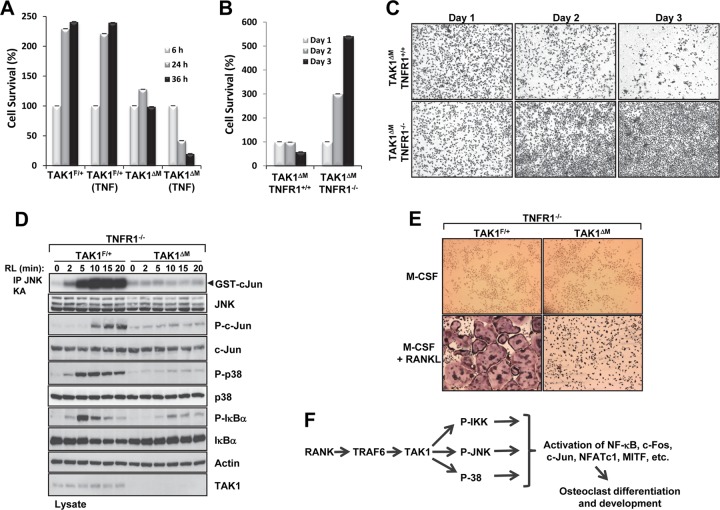
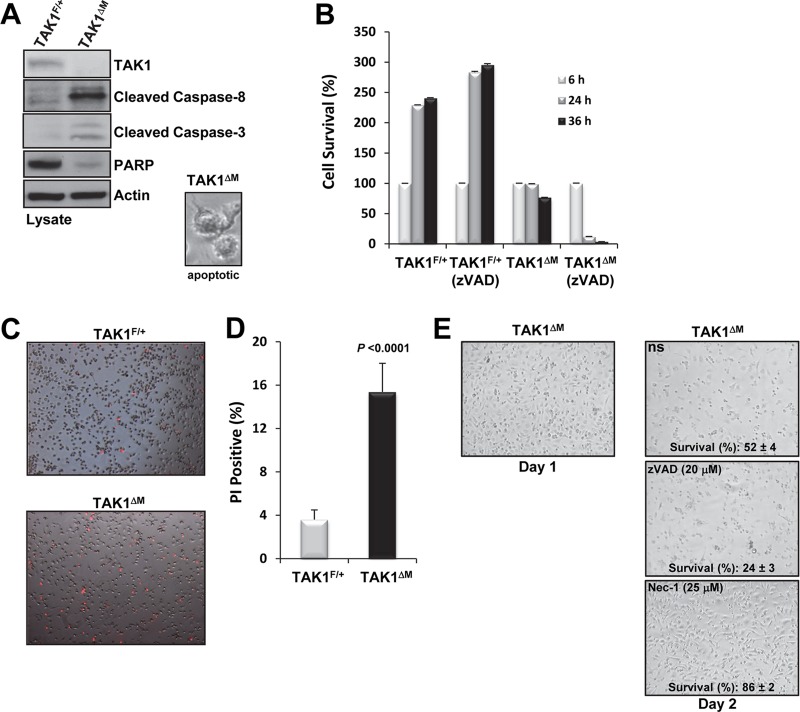
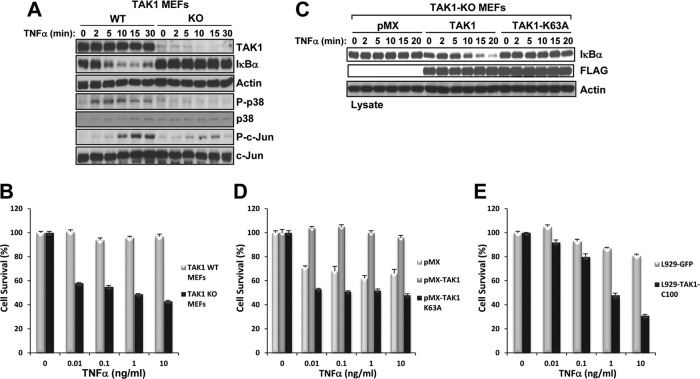
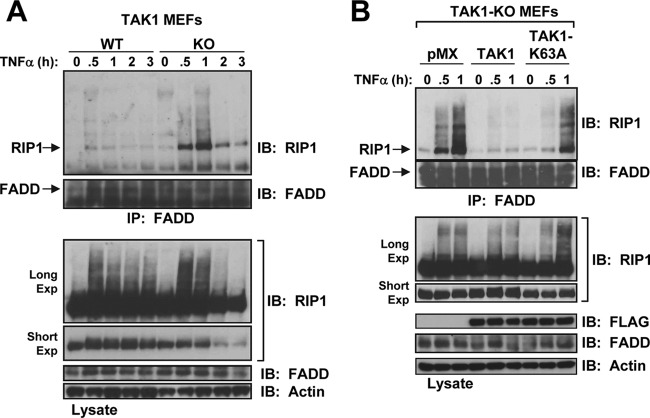
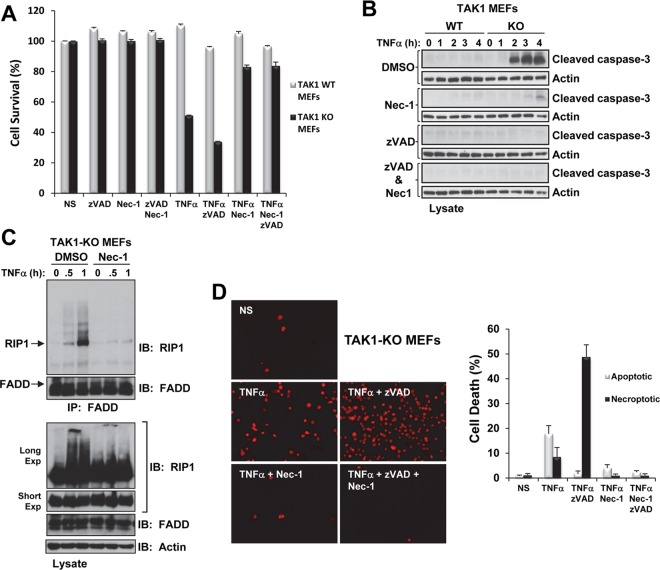
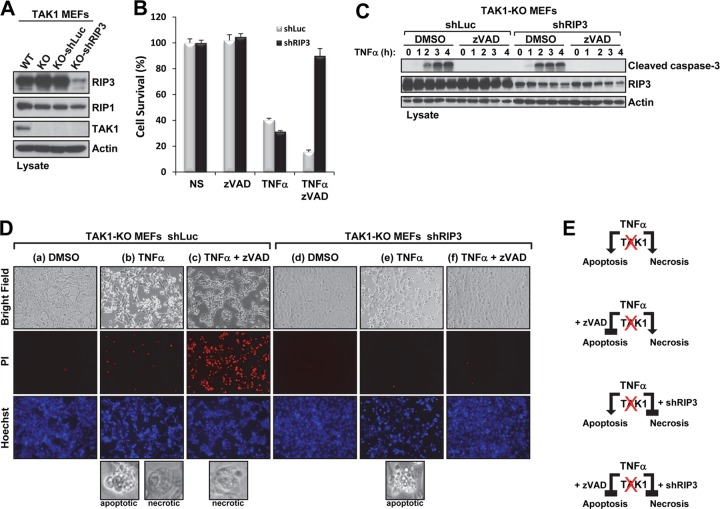
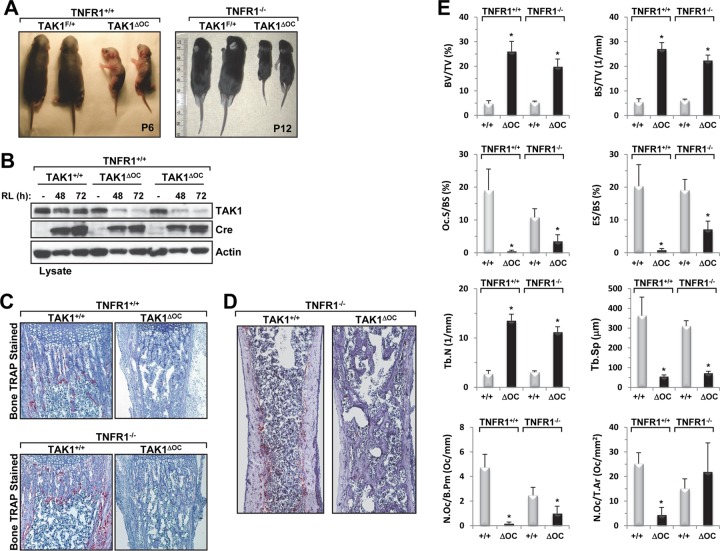
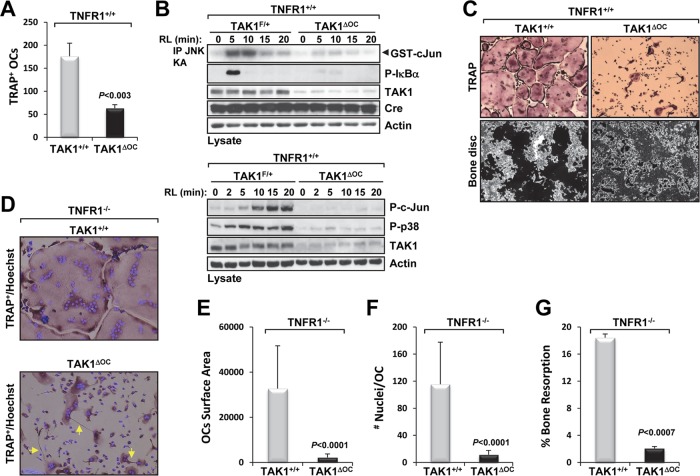
Transforming growth factor β (TGF-β)-activated kinase 1 (TAK1), a mitogen-activated protein 3 (MAP3) kinase, plays an essential role in inflammation by activating the IκB kinase (IKK)/nuclear factor κB (NF-κB) and stress kinase (p38 and c-Jun N-terminal kinase [JNK]) pathways in response to many stimuli. The tumor necrosis factor (TNF) superfamily member receptor activator of NF-κB ligand (RANKL) regulates osteoclastogenesis through its receptor, RANK, and the signaling adaptor TRAF6. Because TAK1 activation is mediated through TRAF6 in the interleukin 1 receptor (IL-1R) and toll-like receptor (TLR) pathways, we sought to investigate the consequence of TAK1 deletion in RANKL-mediated osteoclastogenesis. We generated macrophage colony-stimulating factor (M-CSF)-derived monocytes from the bone marrow of mice with TAK1 deletion in the myeloid lineage. Unexpectedly, TAK1-deficient monocytes in culture died rapidly but could be rescued by retroviral expression of TAK1, inhibition of receptor-interacting protein 1 (RIP1) kinase activity with necrostatin-1, or simultaneous genetic deletion of TNF receptor 1 (TNFR1). Further investigation using TAK1-deficient mouse embryonic fibroblasts revealed that TNF-α-induced cell death was abrogated by the simultaneous inhibition of caspases and knockdown of RIP3, suggesting that TAK1 is an important modulator of both apoptosis and necroptosis. Moreover, TAK1-deficient monocytes rescued from programmed cell death did not form mature osteoclasts in response to RANKL, indicating that TAK1 is indispensable to RANKL-induced osteoclastogenesis. To our knowledge, we are the first to report that mice in which TAK1 has been conditionally deleted in osteoclasts develop osteopetrosis.
Figures
References
-
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J. 1999. RANK is essential for osteoclast and lymph node development. Genes Dev. 13:2412–2424 - PMC - PubMed
-
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323 - PubMed
-
- Darnay BG, Besse A, Poblenz AT, Lamothe B, Jacoby JJ. 2007. TRAFs in RANK signaling. Adv. Exp. Med. Biol. 597:152–159 - PubMed
-
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW. 1999. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13:1015–1024 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous