

ERK1/2 and p38 MAPKs are complementarily involved in estradiol 17ß-D-glucuronide-induced cholestasis: crosstalk with cPKC and PI3K
- PMID: 23166621
- PMCID: PMC3498151
- DOI: 10.1371/journal.pone.0049255
ERK1/2 and p38 MAPKs are complementarily involved in estradiol 17ß-D-glucuronide-induced cholestasis: crosstalk with cPKC and PI3K
Abstract
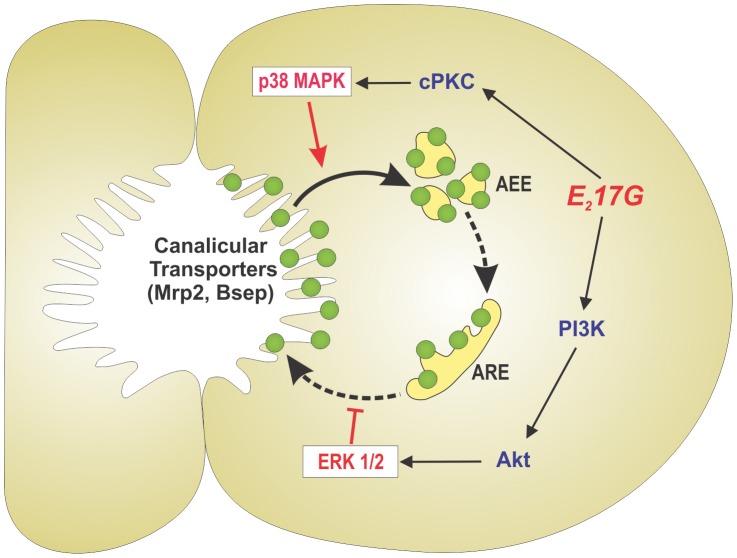
Objective: The endogenous, cholestatic metabolite estradiol 17ß-D-glucuronide (E(2)17G) induces endocytic internalization of the canalicular transporters relevant to bile formation, Bsep and Mrp2. We evaluated here whether MAPKs are involved in this effect.
Design: ERK1/2, JNK1/2, and p38 MAPK activation was assessed by the increase in their phosphorylation status. Hepatocanalicular function was evaluated in isolated rat hepatocyte couplets (IRHCs) by quantifying the apical secretion of fluorescent Bsep and Mrp2 substrates, and in isolated, perfused rat livers (IPRLs), using taurocholate and 2,4-dinitrophenyl-S-glutathione, respectively. Protein kinase participation in E(2)17G-induced secretory failure was assessed by co-administering selective inhibitors. Internalization of Bsep/Mrp2 was assessed by confocal microscopy and image analysis.
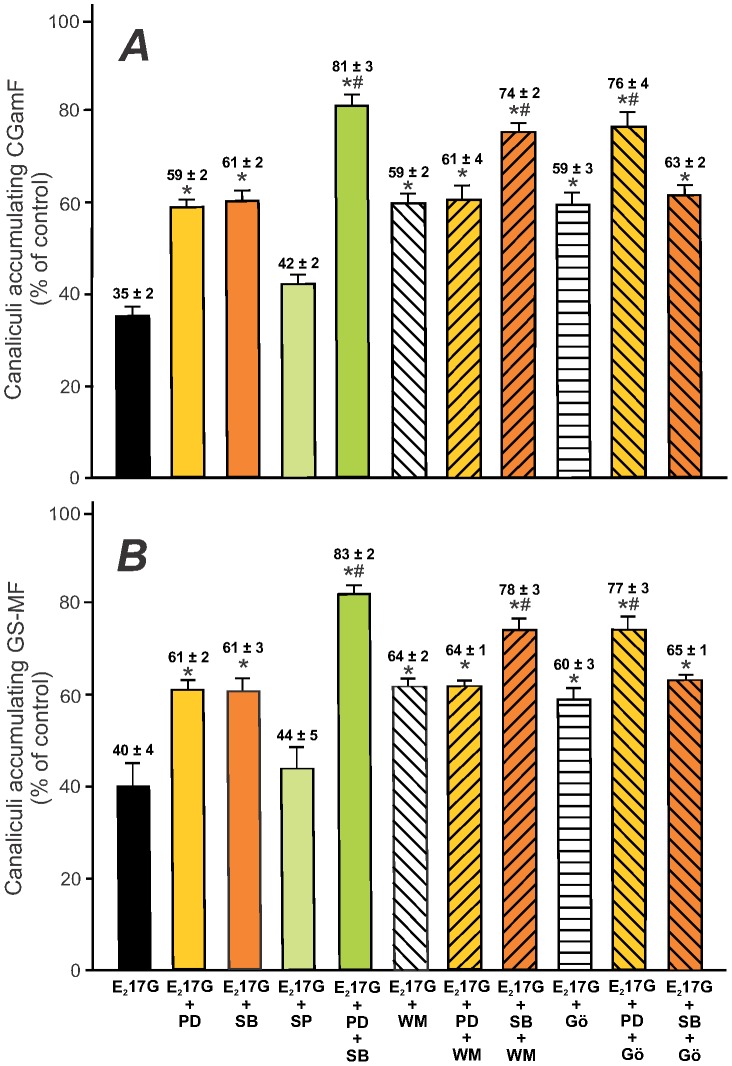
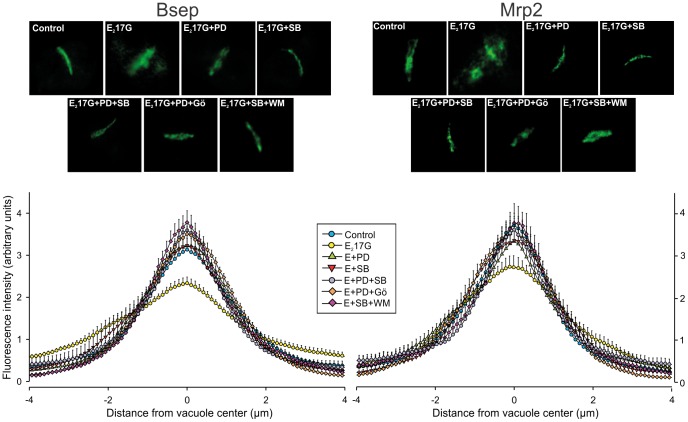
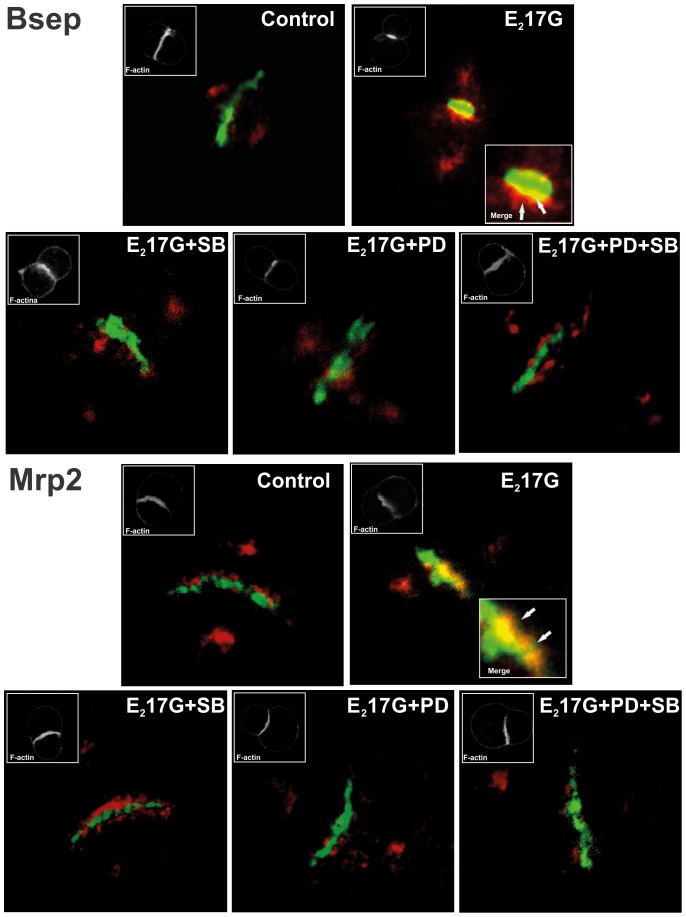
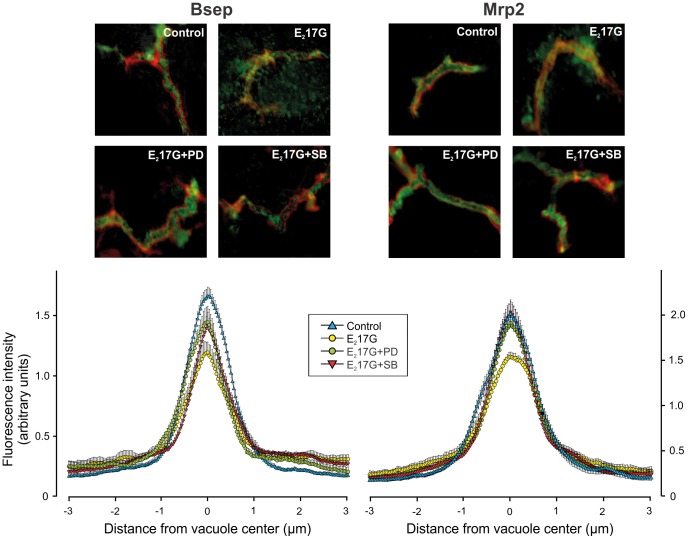
Results: E(2)17G activated all kinds of MAPKs. The PI3K inhibitor wortmannin prevented ERK1/2 activation, whereas the cPKC inhibitor Gö6976 prevented p38 activation, suggesting that ERK1/2 and p38 are downstream of PI3K and cPKC, respectively. The p38 inhibitor SB203580 and the ERK1/2 inhibitor PD98059, but not the JNK1/2 inhibitor SP600125, partially prevented E(2)17G-induced changes in transporter activity and localization in IRHCs. p38 and ERK1/2 co-inhibition resulted in additive protection, suggesting complementary involvement of these MAPKs. In IPRLs, E(2)17G induced endocytosis of canalicular transporters and a rapid and sustained decrease in bile flow and biliary excretion of Bsep/Mrp2 substrates. p38 inhibition prevented this initial decay, and the internalization of Bsep/Mrp2. Contrarily, ERK1/2 inhibition accelerated the recovery of biliary secretion and the canalicular reinsertion of Bsep/Mrp2.
Conclusions: cPKC/p38 MAPK and PI3K/ERK1/2 signalling pathways participate complementarily in E(2)17G-induced cholestasis, through internalization and sustained intracellular retention of canalicular transporters, respectively.
Conflict of interest statement
Figures


References
-
- Crocenzi FA, Zucchetti AE, Boaglio AC, Barosso IR, Sanchez Pozzi EJ, et al. (2012) Localization status of hepatocellular transporters in cholestasis. Front Biosci 17: 1201–1218. - PubMed
-
- Vore M, Liu Y, Huang L (1997) Cholestatic properties and hepatic transport of steroid glucuronides. Drug Metabol Rev 29: 183–203. - PubMed
-
- Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ (2000) Drug- and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology 118: 422–430. - PubMed
-
- Crocenzi FA, Mottino AD, Cao J, Veggi LM, Pozzi EJ, et al. (2003) Estradiol-17β-D-glucuronide induces endocytic internalization of Bsep in rats. Am J Physiol Gastrointest Liver Physiol 285: G449–459. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
