

Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia
- PMID: 23169664
- PMCID: PMC3528580
- DOI: 10.1073/pnas.1215349109
Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia
Abstract
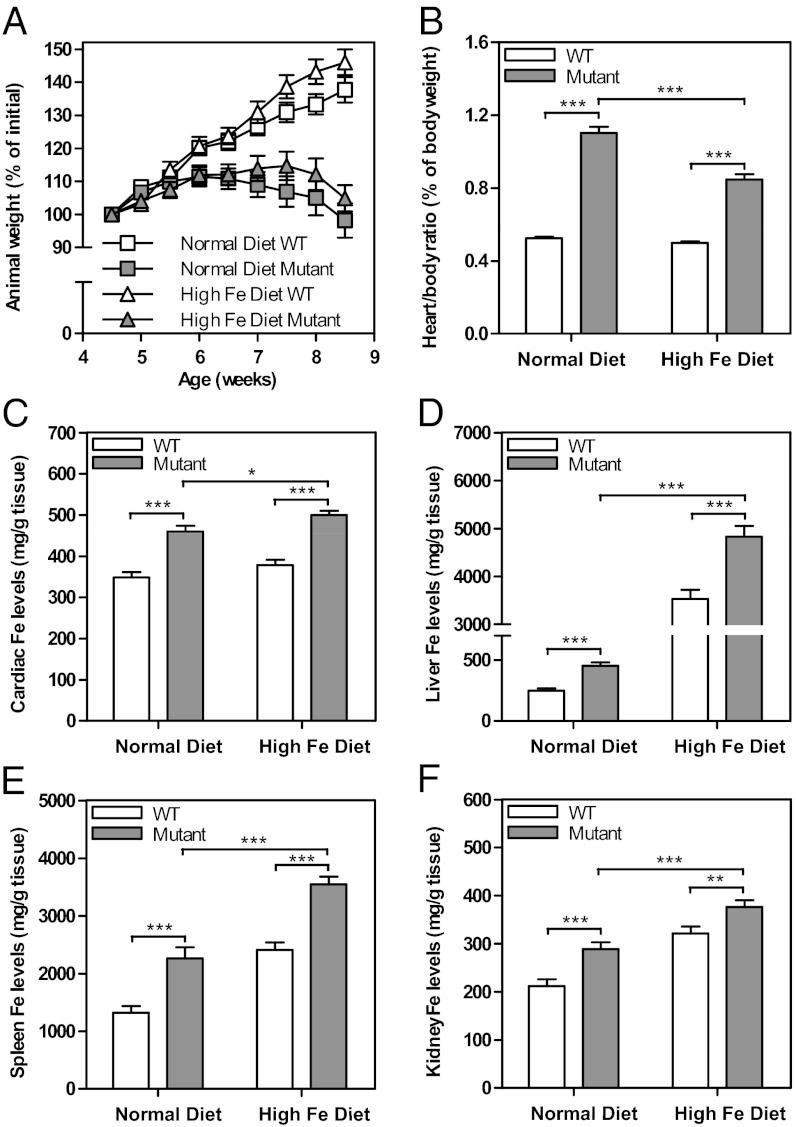
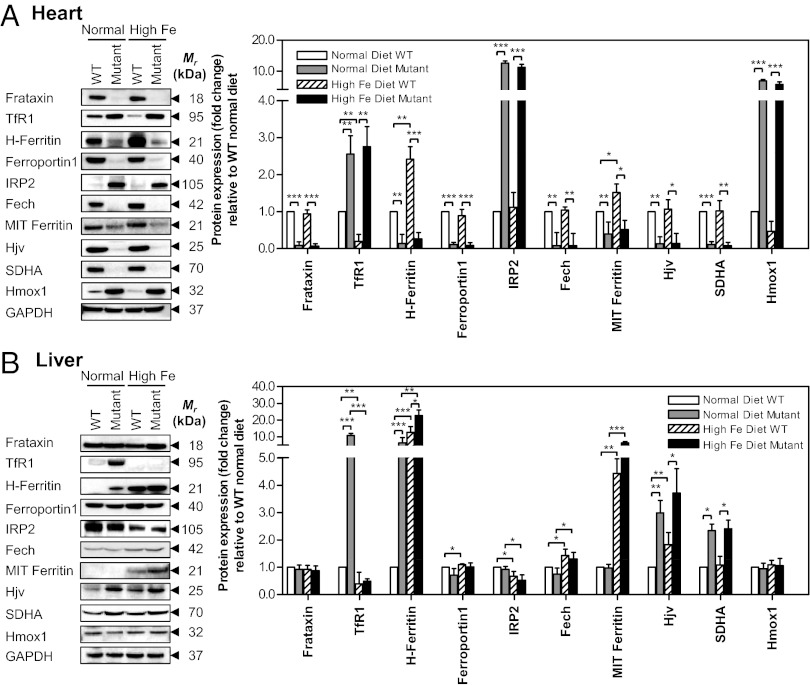
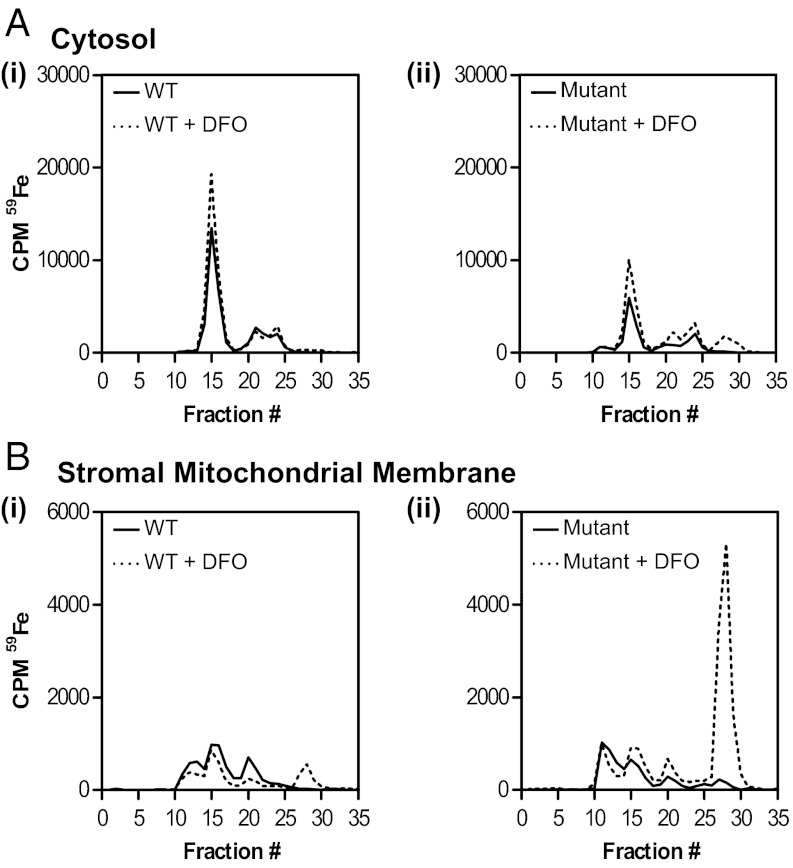
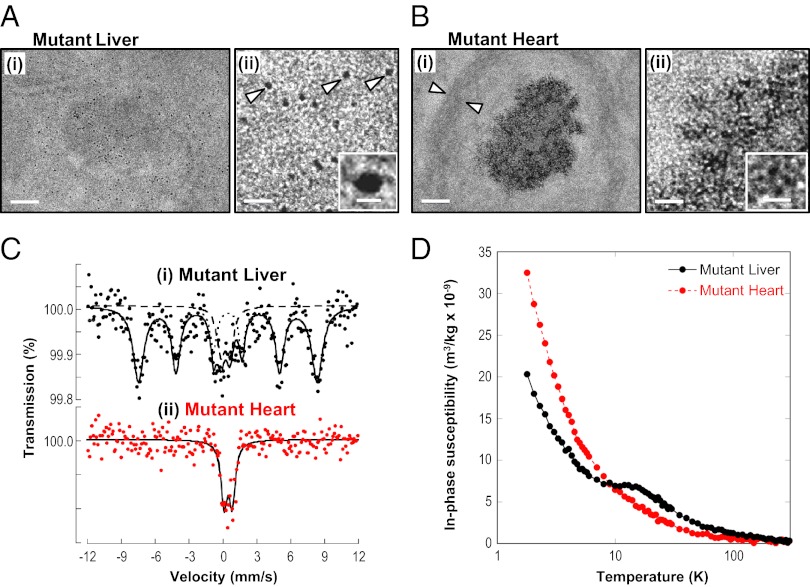
There is no effective treatment for the cardiomyopathy of the most common autosomal recessive ataxia, Friedreich ataxia (FA). This disease is due to decreased expression of the mitochondrial protein, frataxin, which leads to alterations in mitochondrial iron (Fe) metabolism. The identification of potentially toxic mitochondrial Fe deposits in FA suggests Fe plays a role in its pathogenesis. Studies using the muscle creatine kinase (MCK) conditional frataxin knockout mouse that mirrors the disease have demonstrated frataxin deletion alters cardiac Fe metabolism. Indeed, there are pronounced changes in Fe trafficking away from the cytosol to the mitochondrion, leading to a cytosolic Fe deficiency. Considering Fe deficiency can induce apoptosis and cell death, we examined the effect of dietary Fe supplementation, which led to body Fe loading and limited the cardiac hypertrophy in MCK mutants. Furthermore, this study indicates a unique effect of heart and skeletal muscle-specific frataxin deletion on systemic Fe metabolism. Namely, frataxin deletion induces a signaling mechanism to increase systemic Fe levels and Fe loading in tissues where frataxin expression is intact (i.e., liver, kidney, and spleen). Examining the mutant heart, native size-exclusion chromatography, transmission electron microscopy, Mössbauer spectroscopy, and magnetic susceptibility measurements demonstrated that in the absence of frataxin, mitochondria contained biomineral Fe aggregates, which were distinctly different from isolated mammalian ferritin molecules. These mitochondrial aggregates of Fe, phosphorus, and sulfur, probably contribute to the oxidative stress and pathology observed in the absence of frataxin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The MCK mouse heart model of Friedreich's ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation.Proc Natl Acad Sci U S A. 2008 Jul 15;105(28):9757-62. doi: 10.1073/pnas.0804261105. Epub 2008 Jul 9. Proc Natl Acad Sci U S A. 2008. PMID: 18621680 Free PMC article.
-
Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant.Proc Natl Acad Sci U S A. 2009 Sep 22;106(38):16381-6. doi: 10.1073/pnas.0906784106. Epub 2009 Sep 4. Proc Natl Acad Sci U S A. 2009. PMID: 19805308 Free PMC article.
-
The potential of the novel NAD+ supplementing agent, SNH6, as a therapeutic strategy for the treatment of Friedreich's ataxia.Pharmacol Res. 2020 May;155:104680. doi: 10.1016/j.phrs.2020.104680. Epub 2020 Feb 4. Pharmacol Res. 2020. PMID: 32032665
-
Iron metabolism and mitochondrial abnormalities in Friedreich ataxia.Blood Cells Mol Dis. 2002 Nov-Dec;29(3):536-47; discussion 548-52. doi: 10.1006/bcmd.2002.0591. Blood Cells Mol Dis. 2002. PMID: 12547248 Review.
-
Iron and Friedreich ataxia.J Neural Transm Suppl. 2006;(70):143-6. doi: 10.1007/978-3-211-45295-0_22. J Neural Transm Suppl. 2006. PMID: 17017521 Review.
Cited by
-
Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities.Antioxid Redox Signal. 2014 Mar 10;20(8):1324-63. doi: 10.1089/ars.2012.4931. Epub 2013 Aug 15. Antioxid Redox Signal. 2014. PMID: 23815406 Free PMC article. Review.
-
Yeast cells depleted of the frataxin homolog Yfh1 redistribute cellular iron: Studies using Mössbauer spectroscopy and mathematical modeling.J Biol Chem. 2022 Jun;298(6):101921. doi: 10.1016/j.jbc.2022.101921. Epub 2022 Apr 10. J Biol Chem. 2022. PMID: 35413285 Free PMC article.
-
Missense mutations linked to friedreich ataxia have different but synergistic effects on mitochondrial frataxin isoforms.J Biol Chem. 2013 Feb 8;288(6):4116-27. doi: 10.1074/jbc.M112.435263. Epub 2012 Dec 26. J Biol Chem. 2013. PMID: 23269675 Free PMC article.
-
Evolution of an Iron-Detoxifying Protein: Eukaryotic and Rickettsia Frataxins Contain a Conserved Site Which Is Not Present in Their Bacterial Homologues.Int J Mol Sci. 2022 Oct 29;23(21):13151. doi: 10.3390/ijms232113151. Int J Mol Sci. 2022. PMID: 36361939 Free PMC article.
-
Fixing frataxin: 'ironing out' the metabolic defect in Friedreich's ataxia.Br J Pharmacol. 2014 Apr;171(8):2174-90. doi: 10.1111/bph.12470. Br J Pharmacol. 2014. PMID: 24138602 Free PMC article. Review.
References
-
- Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood. 2005;105(5):1867–1874. - PubMed
-
- Puccio H, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27(2):181–186. - PubMed
-
- Lesuisse E, et al. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1) Hum Mol Genet. 2003;12(8):879–889. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
