

Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans
- PMID: 23170002
- PMCID: PMC3509437
- DOI: 10.1128/mBio.00473-12
Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans
Abstract
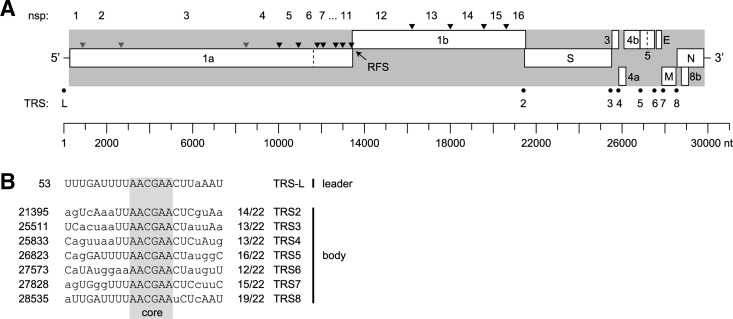
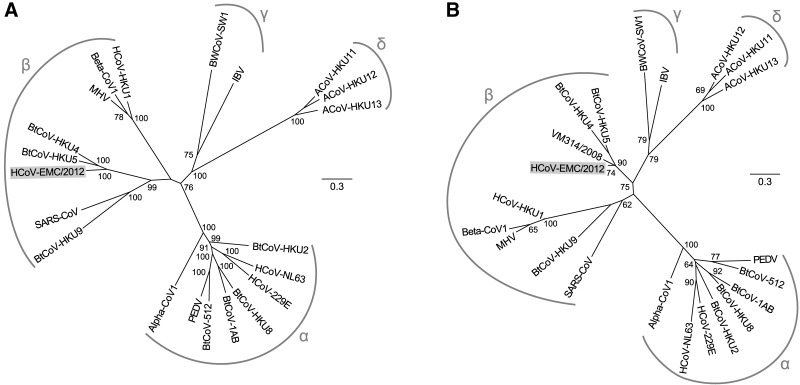
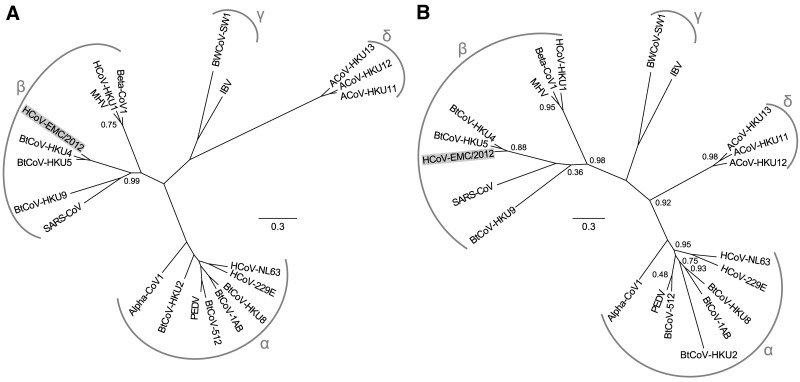
A novel human coronavirus (HCoV-EMC/2012) was isolated from a man with acute pneumonia and renal failure in June 2012. This report describes the complete genome sequence, genome organization, and expression strategy of HCoV-EMC/2012 and its relation with known coronaviruses. The genome contains 30,119 nucleotides and contains at least 10 predicted open reading frames, 9 of which are predicted to be expressed from a nested set of seven subgenomic mRNAs. Phylogenetic analysis of the replicase gene of coronaviruses with completely sequenced genomes showed that HCoV-EMC/2012 is most closely related to Tylonycteris bat coronavirus HKU4 (BtCoV-HKU4) and Pipistrellus bat coronavirus HKU5 (BtCoV-HKU5), which prototype two species in lineage C of the genus Betacoronavirus. In accordance with the guidelines of the International Committee on Taxonomy of Viruses, and in view of the 75% and 77% amino acid sequence identity in 7 conserved replicase domains with BtCoV-HKU4 and BtCoV-HKU5, respectively, we propose that HCoV-EMC/2012 prototypes a novel species in the genus Betacoronavirus. HCoV-EMC/2012 may be most closely related to a coronavirus detected in Pipistrellus pipistrellus in The Netherlands, but because only a short sequence from the most conserved part of the RNA-dependent RNA polymerase-encoding region of the genome was reported for this bat virus, its genetic distance from HCoV-EMC remains uncertain. HCoV-EMC/2012 is the sixth coronavirus known to infect humans and the first human virus within betacoronavirus lineage C.
Importance: Coronaviruses are capable of infecting humans and many animal species. Most infections caused by human coronaviruses are relatively mild. However, the outbreak of severe acute respiratory syndrome (SARS) caused by SARS-CoV in 2002 to 2003 and the fatal infection of a human by HCoV-EMC/2012 in 2012 show that coronaviruses are able to cause severe, sometimes fatal disease in humans. We have determined the complete genome of HCoV-EMC/2012 using an unbiased virus discovery approach involving next-generation sequencing techniques, which enabled subsequent state-of-the-art bioinformatics, phylogenetics, and taxonomic analyses. By establishing its complete genome sequence, HCoV-EMC/2012 was characterized as a new genotype which is closely related to bat coronaviruses that are distant from SARS-CoV. We expect that this information will be vital to rapid advancement of both clinical and vital research on this emerging pathogen.
Figures
Comment in
-
The new age of virus discovery: genomic analysis of a novel human betacoronavirus isolated from a fatal case of pneumonia.mBio. 2013 Jan 8;4(1):e00548-12. doi: 10.1128/mBio.00548-12. mBio. 2013. PMID: 23300251 Free PMC article.
References
-
- de Groot RJ, et al. 2012. Family Coronaviridae, p. 806–828 In King AMQ, Adams MJ, Cartens EB, Lefkowitz EJ, Virus taxonomy, the 9th report of the international committee on taxonomy of viruses. Academic Press, San Diego, CA.
-
- Li W, et al. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
