

Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus
- PMID: 23173743
- PMCID: PMC3575255
- DOI: 10.1186/1750-1326-7-59
Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus
Abstract
Background: Axonal pathology might constitute one of the earliest manifestations of Alzheimer disease. Axonal dystrophies were observed in Alzheimer's patients and transgenic models at early ages. These axonal dystrophies could reflect the disruption of axonal transport and the accumulation of multiple vesicles at local points. It has been also proposed that dystrophies might interfere with normal intracellular proteolysis. In this work, we have investigated the progression of the hippocampal pathology and the possible implication in Abeta production in young (6 months) and aged (18 months) PS1(M146L)/APP(751sl) transgenic mice.
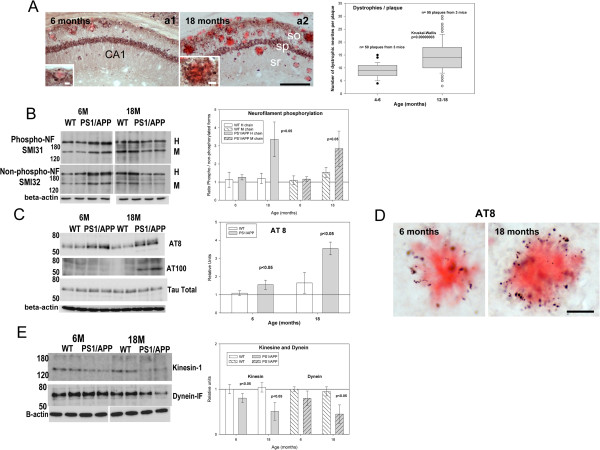
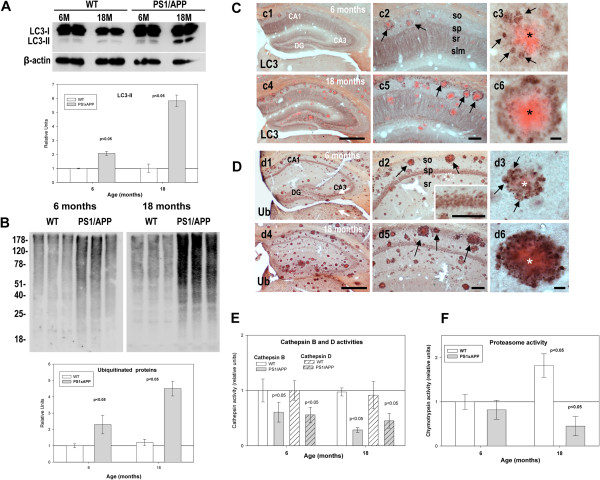
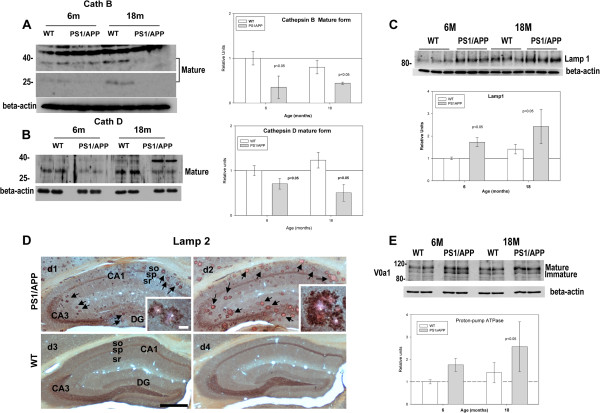
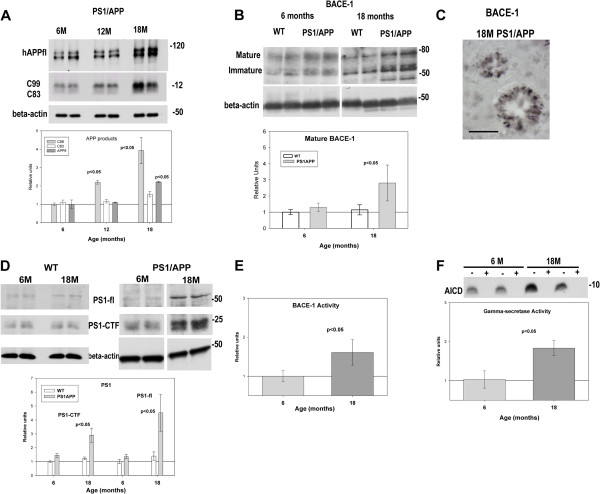
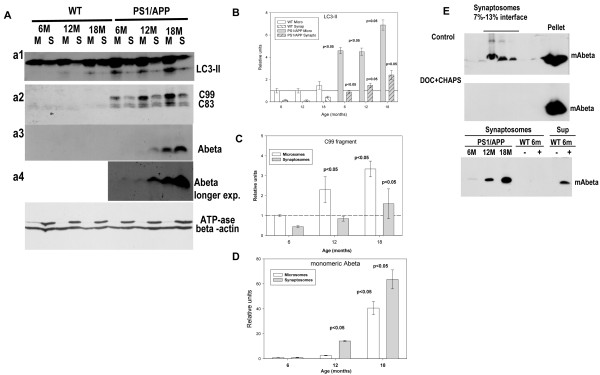
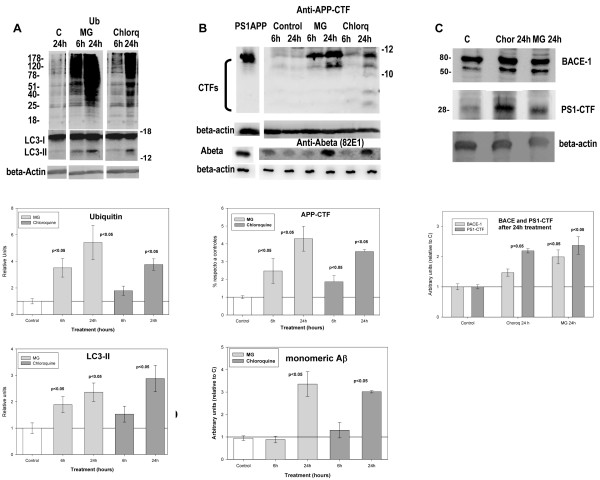
Results: Our data demonstrated the existence of a progressive, age-dependent, formation of axonal dystrophies, mainly located in contact with congophilic Abeta deposition, which exhibited tau and neurofilament hyperphosphorylation. This progressive pathology was paralleled with decreased expression of the motor proteins kinesin and dynein. Furthermore, we also observed an early decrease in the activity of cathepsins B and D, progressing to a deep inhibition of these lysosomal proteases at late ages. This lysosomal impairment could be responsible for the accumulation of LC3-II and ubiquitinated proteins within axonal dystrophies. We have also investigated the repercussion of these deficiencies on the APP metabolism. Our data demonstrated the existence of an increase in the amyloidogenic pathway, which was reflected by the accumulation of hAPPfl, C99 fragment, intracellular Abeta in parallel with an increase in BACE and gamma-secretase activities. In vitro experiments, using APPswe transfected N2a cells, demonstrated that any imbalance on the proteolytic systems reproduced the in vivo alterations in APP metabolism. Finally, our data also demonstrated that Abeta peptides were preferentially accumulated in isolated synaptosomes.
Conclusion: A progressive age-dependent cytoskeletal pathology along with a reduction of lysosomal and, in minor extent, proteasomal activity could be directly implicated in the progressive accumulation of APP derived fragments (and Abeta peptides) in parallel with the increase of BACE-1 and gamma-secretase activities. This retard in the APP metabolism seemed to be directly implicated in the synaptic Abeta accumulation and, in consequence, in the pathology progression between synaptically connected regions.
Figures
Similar articles
-
Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer's mice hippocampus.Acta Neuropathol. 2012 Jan;123(1):53-70. doi: 10.1007/s00401-011-0896-x. Epub 2011 Oct 22. Acta Neuropathol. 2012. PMID: 22020633 Free PMC article.
-
Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice.J Neurosci. 2002 Nov 15;22(22):9785-93. doi: 10.1523/JNEUROSCI.22-22-09785.2002. J Neurosci. 2002. PMID: 12427834 Free PMC article.
-
Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer's disease.Graefes Arch Clin Exp Ophthalmol. 2009 Sep;247(9):1213-21. doi: 10.1007/s00417-009-1060-3. Epub 2009 Mar 7. Graefes Arch Clin Exp Ophthalmol. 2009. PMID: 19271231
-
APP transgenic modeling of Alzheimer's disease: mechanisms of neurodegeneration and aberrant neurogenesis.Brain Struct Funct. 2010 Mar;214(2-3):111-26. doi: 10.1007/s00429-009-0232-6. Epub 2009 Nov 29. Brain Struct Funct. 2010. PMID: 20091183 Free PMC article. Review.
-
Alzheimer's disease.Subcell Biochem. 2012;65:329-52. doi: 10.1007/978-94-007-5416-4_14. Subcell Biochem. 2012. PMID: 23225010 Review.
Cited by
-
Acidifying Endolysosomes Prevented Low-Density Lipoprotein-Induced Amyloidogenesis.J Alzheimers Dis. 2019;67(1):393-410. doi: 10.3233/JAD-180941. J Alzheimers Dis. 2019. PMID: 30594929 Free PMC article.
-
Axonal transport of late endosomes and amphisomes is selectively modulated by local Ca2+ efflux and disrupted by PSEN1 loss of function.Sci Adv. 2022 Apr 29;8(17):eabj5716. doi: 10.1126/sciadv.abj5716. Epub 2022 Apr 29. Sci Adv. 2022. PMID: 35486730 Free PMC article.
-
The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease.Front Aging Neurosci. 2014 May 20;6:93. doi: 10.3389/fnagi.2014.00093. eCollection 2014. Front Aging Neurosci. 2014. PMID: 24904407 Free PMC article. Review.
-
DNLA Delayed the Appearance of Learning and Memory Impairment of APP/PS1 Mice: Involvement of mTOR/TFEB/v-ATPase Signaling Pathway.CNS Neurosci Ther. 2025 Mar;31(3):e70300. doi: 10.1111/cns.70300. CNS Neurosci Ther. 2025. PMID: 40047153 Free PMC article.
-
Subcellular proteomics and iPSC modeling uncover reversible mechanisms of axonal pathology in Alzheimer's disease.Nat Aging. 2025 Mar;5(3):504-527. doi: 10.1038/s43587-025-00823-3. Epub 2025 Mar 10. Nat Aging. 2025. PMID: 40065072 Free PMC article.
References
-
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A. et al.Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
