

The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease
- PMID: 23175838
- PMCID: PMC3568961
- DOI: 10.1523/JNEUROSCI.1858-12.2012
The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer's disease
Retraction in
-
Retraction: Larson et al., "The Complex PrPc-Fyn Couples Human Oligomeric Aβ with Pathological Tau Changes in Alzheimer's Disease".J Neurosci. 2025 Mar 12;45(11):e0301252025. doi: 10.1523/JNEUROSCI.0301-25.2025. J Neurosci. 2025. PMID: 40050120 Free PMC article. No abstract available.
Abstract
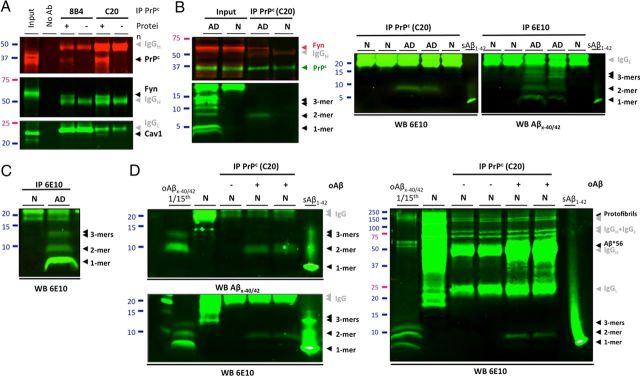
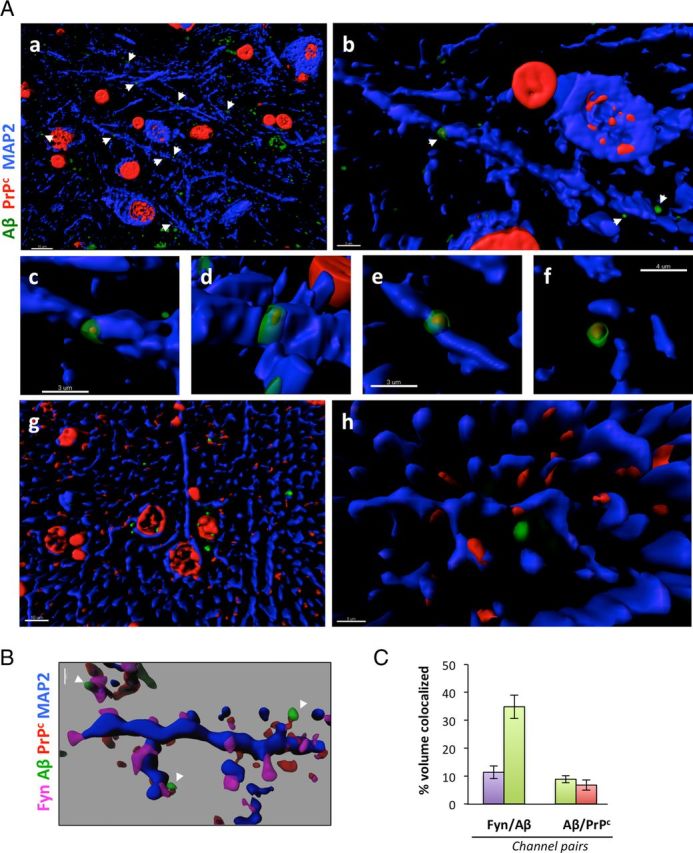
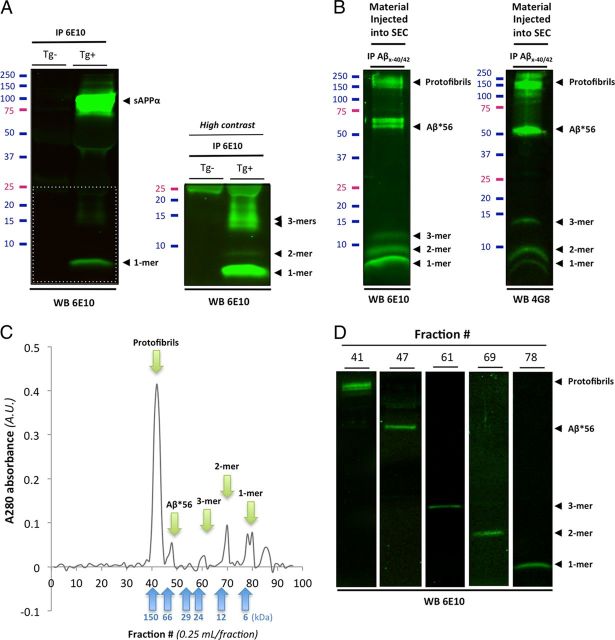
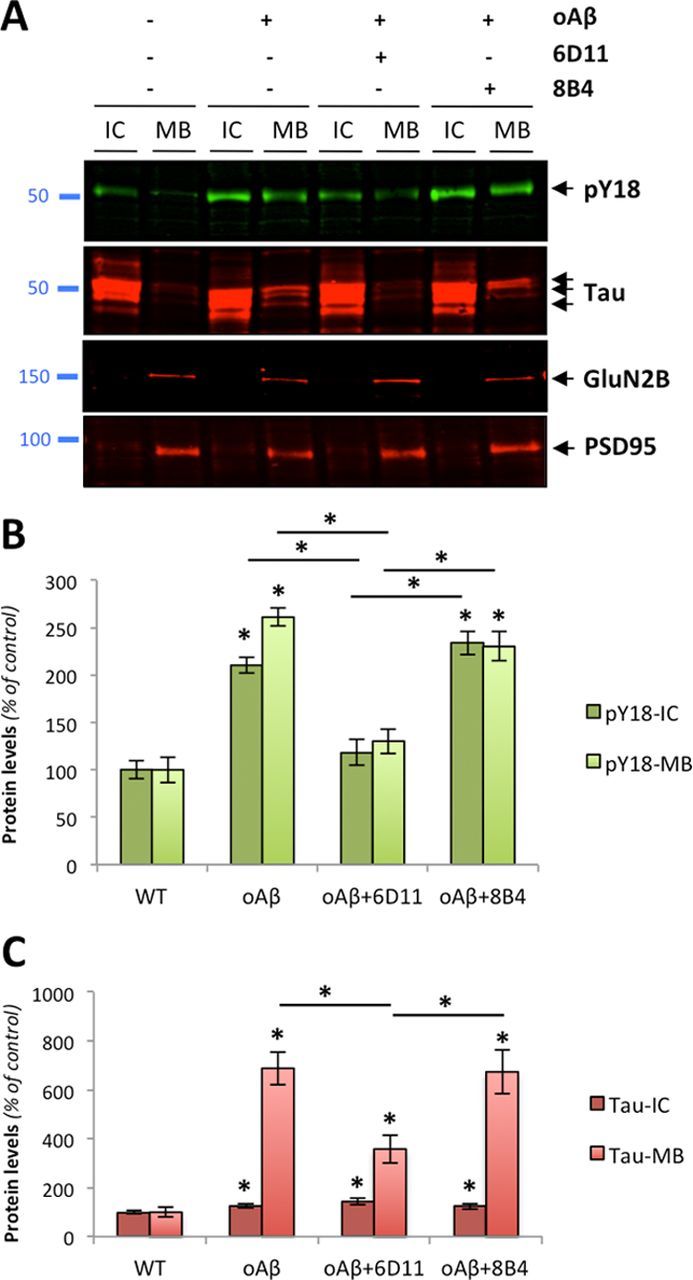
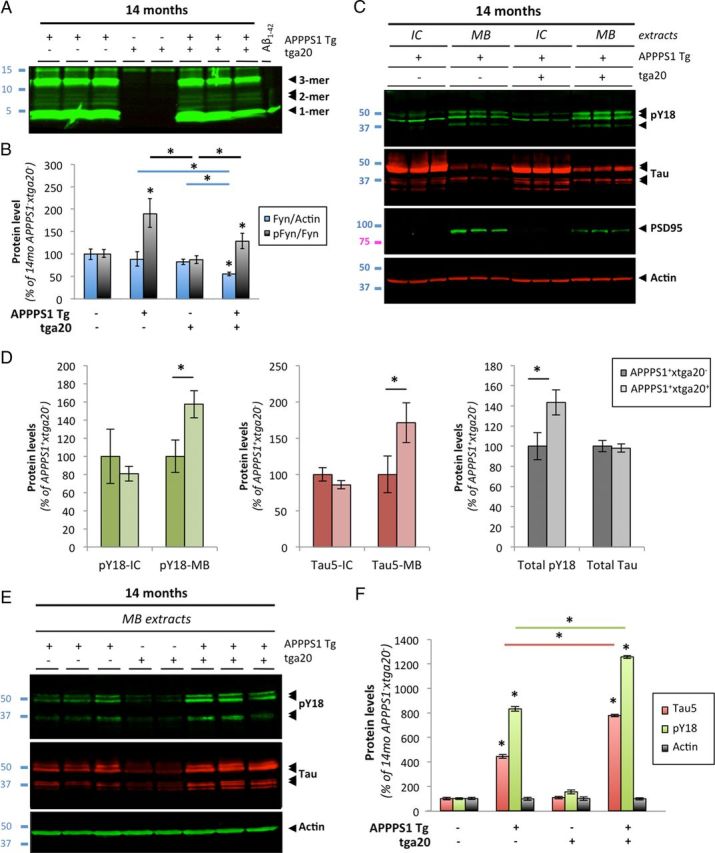
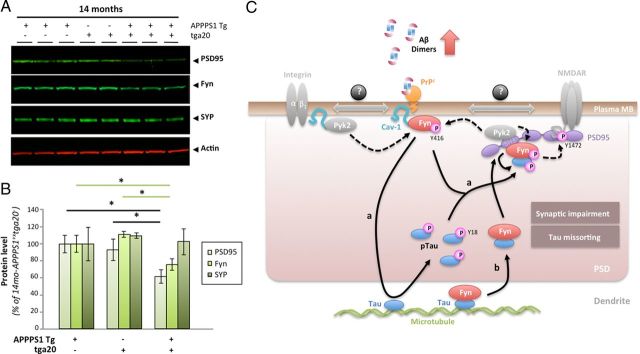
Amid controversy, the cellular form of the prion protein PrP(c) has been proposed to mediate oligomeric amyloid-β (Aβ)-induced deficits. In contrast, there is consistent evidence that the Src kinase Fyn is activated by Aβ oligomers and leads to synaptic and cognitive impairment in transgenic animals. However, the molecular mechanism by which soluble Aβ activates Fyn remains unknown. Combining the use of human and transgenic mouse brain tissue as well as primary cortical neurons, we demonstrate that soluble Aβ binds to PrP(c) at neuronal dendritic spines in vivo and in vitro where it forms a complex with Fyn, resulting in the activation of the kinase. Using the antibody 6D11 to prevent oligomeric Aβ from binding to PrP(c), we abolished Fyn activation and Fyn-dependent tau hyperphosphorylation induced by endogenous oligomeric Aβ in vitro. Finally, we showed that gene dosage of Prnp regulates Aβ-induced Fyn/tau alterations. Together, our findings identify a complete signaling cascade linking one specific endogenous Aβ oligomer, Fyn alteration, and tau hyperphosphorylation in cellular and animal models modeling aspects of the molecular pathogenesis of Alzheimer's disease.
Figures




References
-
- State of aggregation. Nat Neurosci. 2011;14:399. [No authors listed] - PubMed
-
- Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-β oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. - PMC - PubMed
-
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous