

Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation
- PMID: 23178126
- PMCID: PMC3567437
- DOI: 10.1016/j.cell.2012.10.043
Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation
Abstract
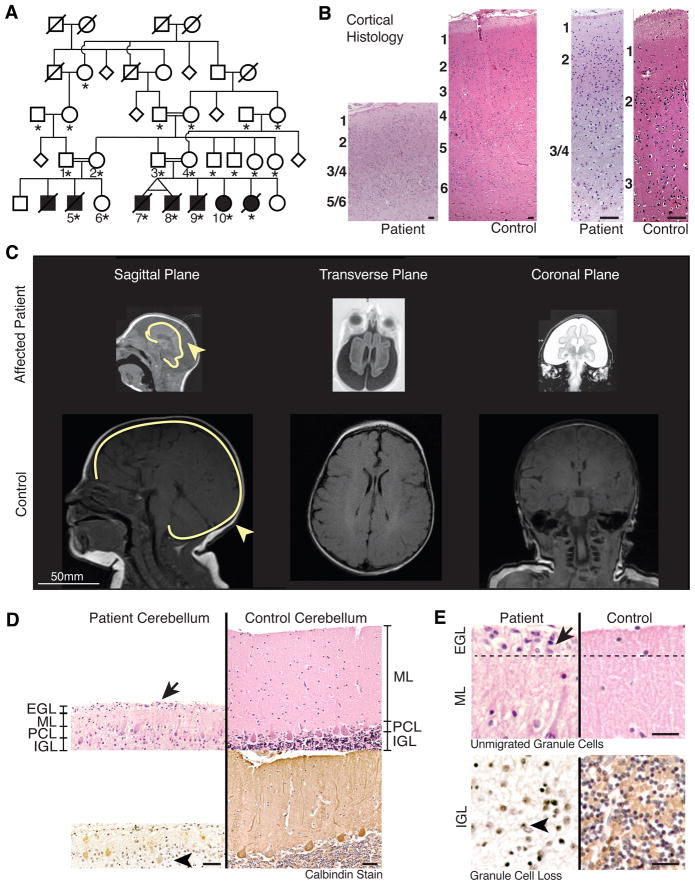
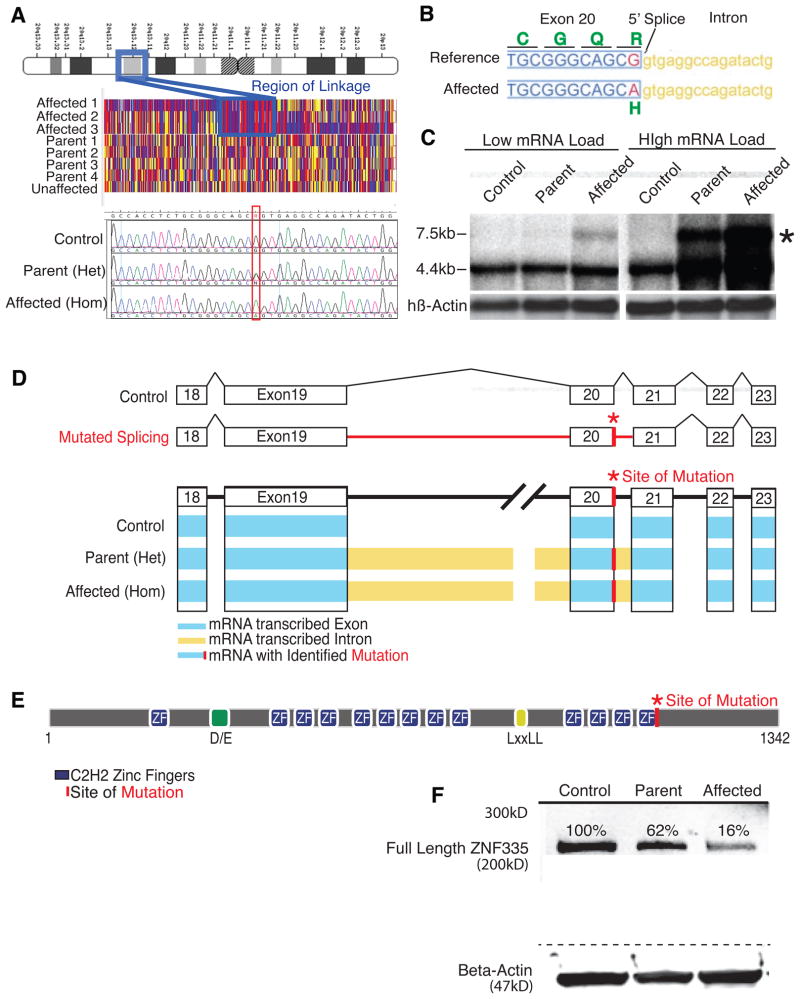
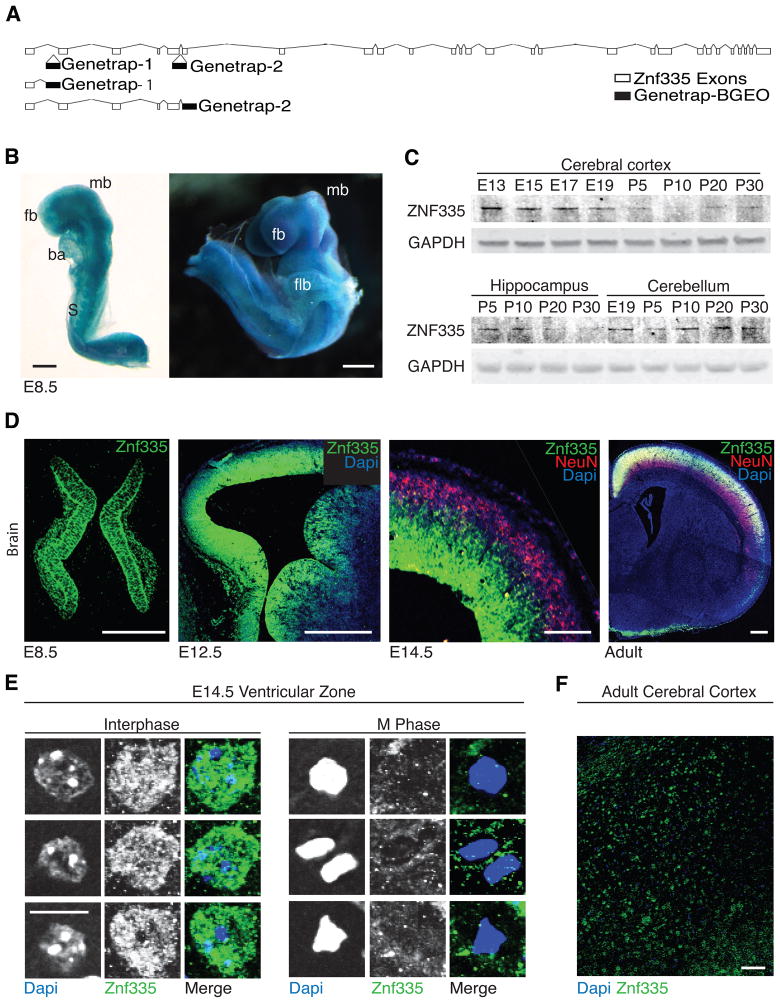
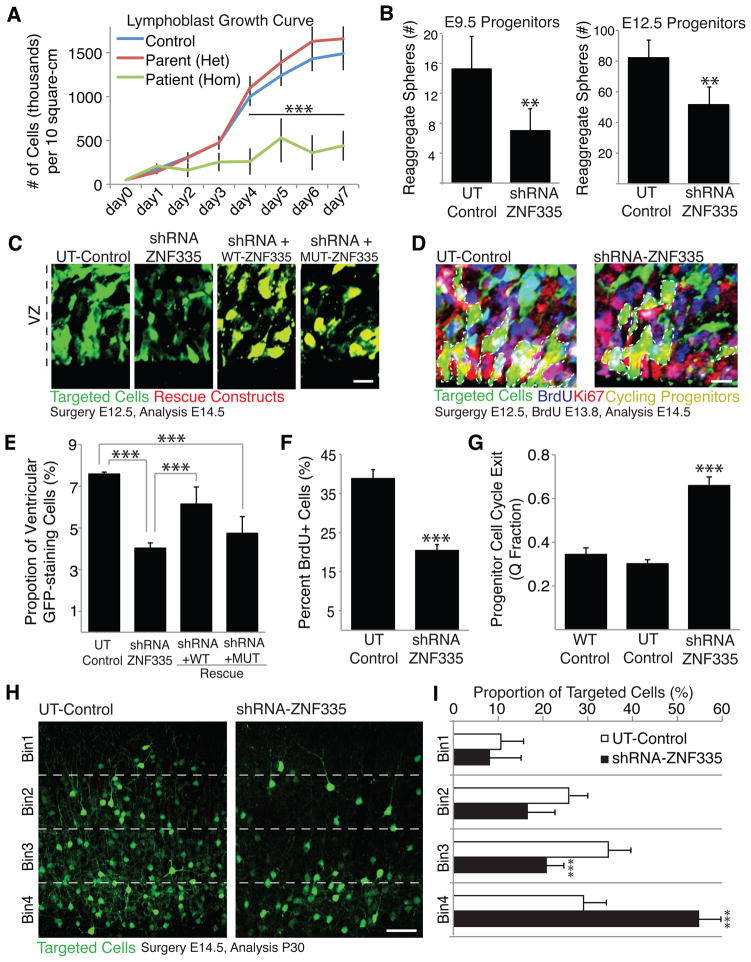
Microcephaly is a neurodevelopmental disorder causing significantly reduced cerebral cortex size. Many known microcephaly gene products localize to centrosomes, regulating cell fate and proliferation. Here, we identify and characterize a nuclear zinc finger protein, ZNF335/NIF-1, as a causative gene for severe microcephaly, small somatic size, and neonatal death. Znf335 null mice are embryonically lethal, and conditional knockout leads to severely reduced cortical size. RNA-interference and postmortem human studies show that ZNF335 is essential for neural progenitor self-renewal, neurogenesis, and neuronal differentiation. ZNF335 is a component of a vertebrate-specific, trithorax H3K4-methylation complex, directly regulating REST/NRSF, a master regulator of neural gene expression and cell fate, as well as other essential neural-specific genes. Our results reveal ZNF335 as an essential link between H3K4 complexes and REST/NRSF and provide the first direct genetic evidence that this pathway regulates human neurogenesis and neuronal differentiation.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. - PubMed
-
- Barkovich AJ, Moore KR, Jones BV, Vezina G, Koch BL, Raybaud C, Grant PE, Blaser SI, Hedlund GL, Illner A. Diagnostic Imaging: Pediatric Neuroradiology. 1. Salt Lake City, Utah: Amirsys- Elsevier; 2007.
-
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
- R01 GM071004/GM/NIGMS NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R01 CA118487/CA/NCI NIH HHS/United States
- R01 DK016636/DK/NIDDK NIH HHS/United States
- GM058012/GM/NIGMS NIH HHS/United States
- T32 HD007466/HD/NICHD NIH HHS/United States
- T32GM007753/GM/NIGMS NIH HHS/United States
- GM071004/GM/NIGMS NIH HHS/United States
- R01 NS032457/NS/NINDS NIH HHS/United States
- R01 NS35129/NS/NINDS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 DK16636/DK/NIDDK NIH HHS/United States
- R01 NS035129/NS/NINDS NIH HHS/United States
- CA118487/CA/NCI NIH HHS/United States
- R01 GM058012/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
