

Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma
- PMID: 23178492
- PMCID: PMC3899696
- DOI: 10.1038/onc.2012.525
Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma
Erratum in
-
Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma.Oncogene. 2016 Nov 24;35(47):6155-6156. doi: 10.1038/onc.2016.142. Epub 2016 Jun 27. Oncogene. 2016. PMID: 27345405 No abstract available.
Abstract
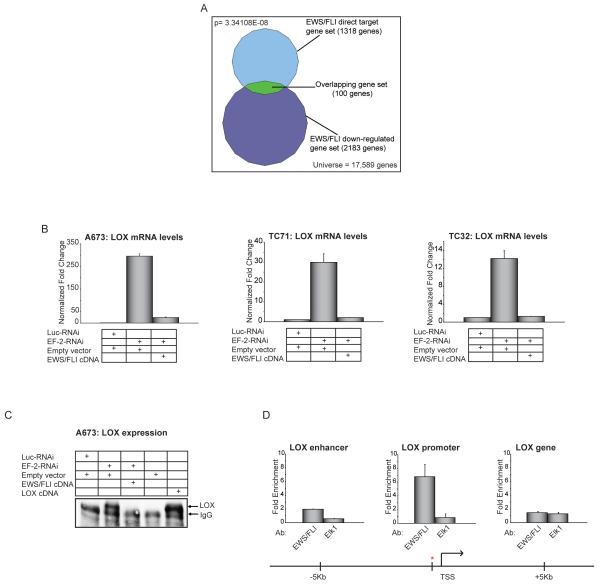
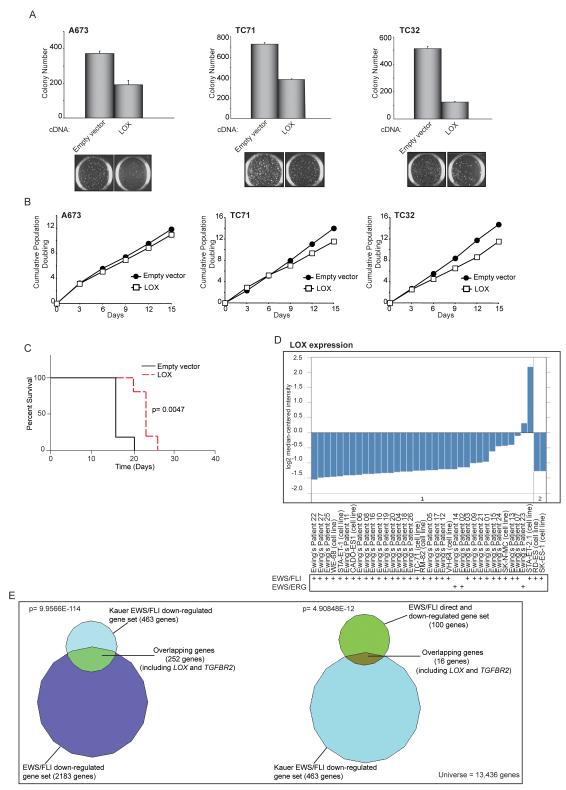
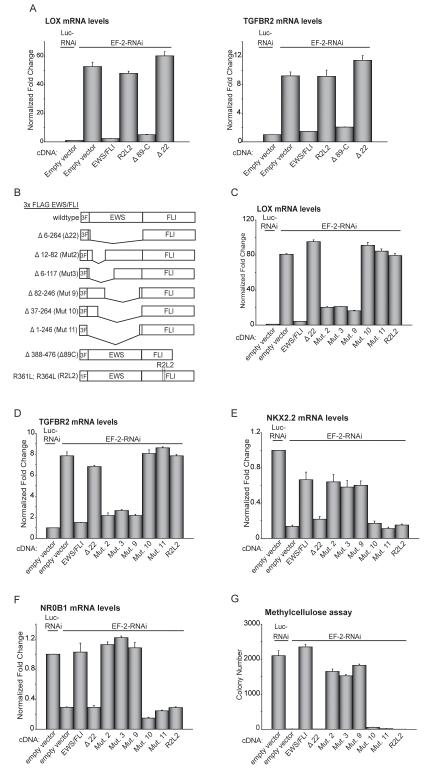
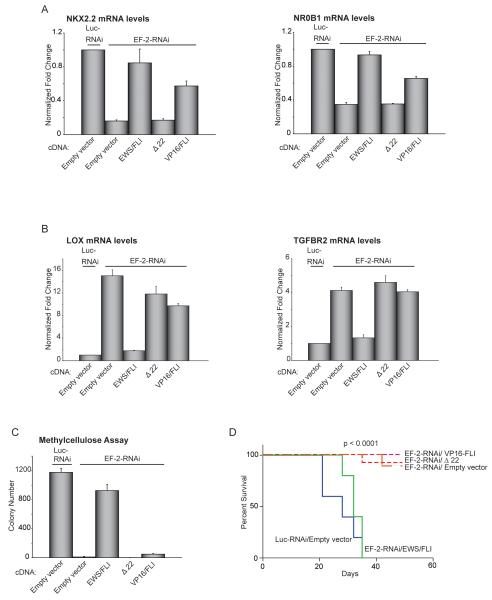
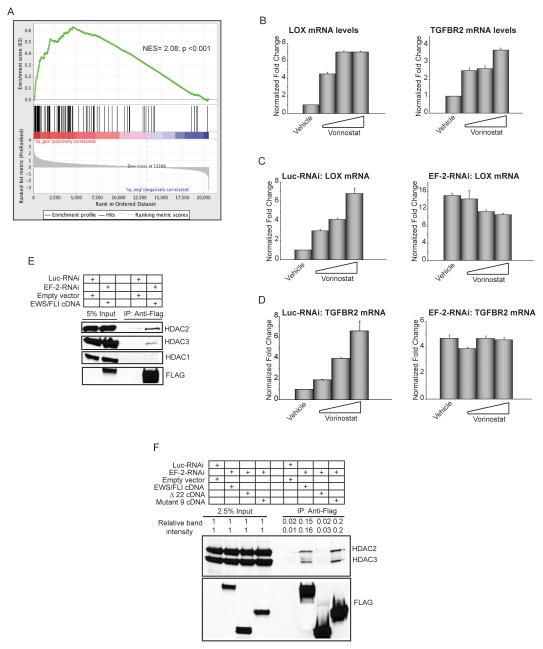
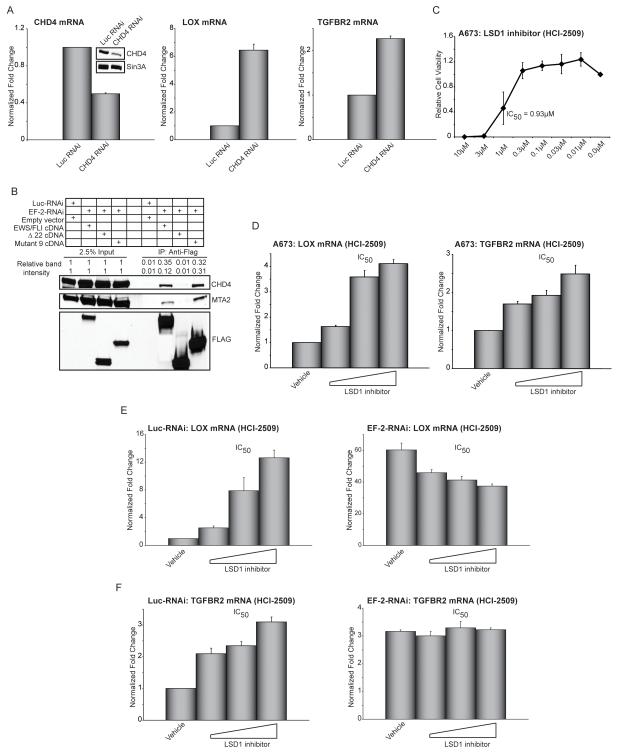
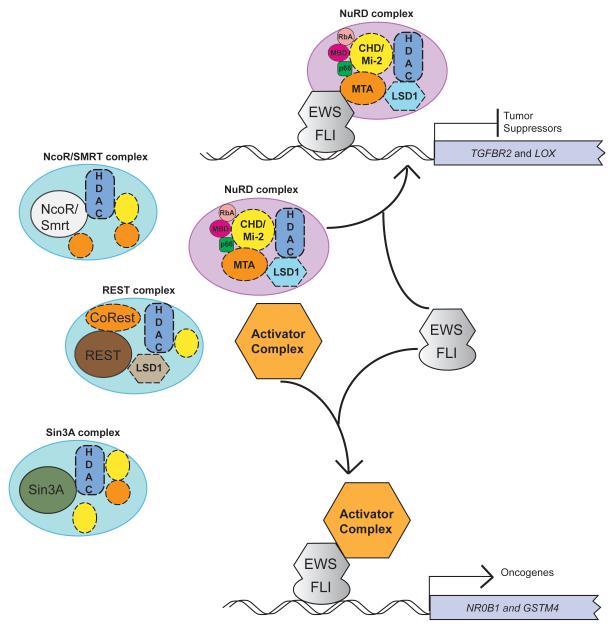
Ewing sarcoma provides an important model for transcription-factor-mediated oncogenic transformation because of its reliance on the ETS-type fusion oncoprotein EWS/FLI. EWS/FLI functions as a transcriptional activator and transcriptional activation is required for its oncogenic activity. Here, we demonstrate that a previously less-well characterized transcriptional repressive function of the EWS/FLI fusion is also required for the transformed phenotype of Ewing sarcoma. Through comparison of EWS/FLI transcriptional profiling and genome-wide localization data, we define the complement of EWS/FLI direct downregulated target genes. We demonstrate that LOX is a previously undescribed EWS/FLI-repressed target that inhibits the transformed phenotype of Ewing sarcoma cells. Mechanistic studies demonstrate that the NuRD co-repressor complex interacts with EWS/FLI, and that its associated histone deacetylase and LSD1 activities contribute to the repressive function. Taken together, these data reveal a previously unknown molecular function for EWS/FLI, demonstrate a more highly coordinated oncogenic transcriptional hierarchy mediated by EWS/FLI than previously suspected, and implicate a new paradigm for therapeutic intervention aimed at controlling NuRD activity in Ewing sarcoma tumors.
Figures
References
-
- Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–165. - PubMed
-
- Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer. 2005;41:2462–2478. - PubMed
-
- Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene. 1998;17:603–610. - PubMed
-
- Ramakrishnan R, Fujimura Y, Zou JP, Liu F, Lee L, Rao VN, et al. Role of protein-protein interactions in the antiapoptotic function of EWS-Fli-1. Oncogene. 2004;23:7087–7094. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
