

The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction
- PMID: 23185029
- PMCID: PMC3514785
- DOI: 10.1083/jcb.201208152
The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction
Abstract
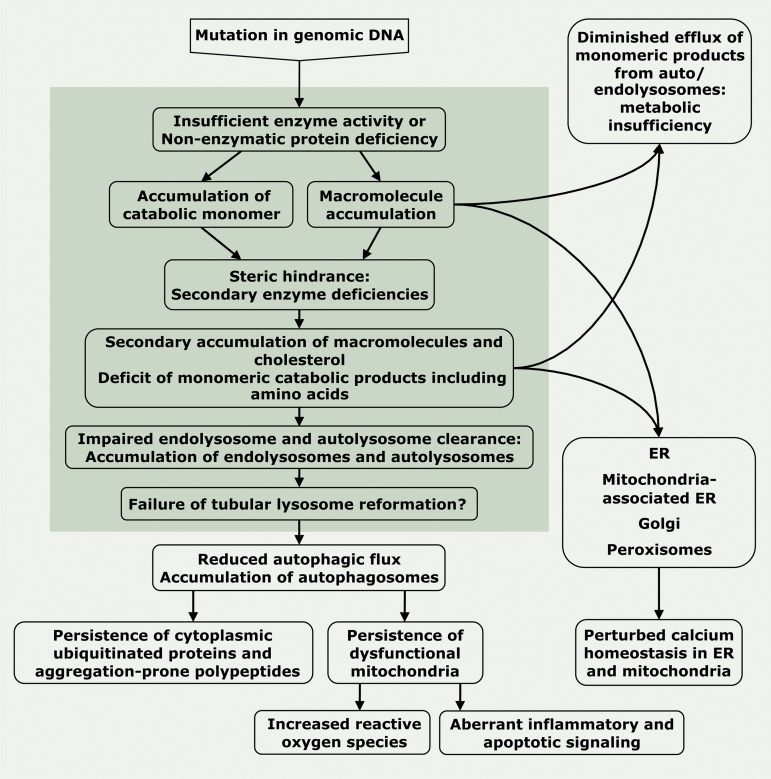
Lysosomal storage diseases (LSDs) are a family of disorders that result from inherited gene mutations that perturb lysosomal homeostasis. LSDs mainly stem from deficiencies in lysosomal enzymes, but also in some non-enzymatic lysosomal proteins, which lead to abnormal storage of macromolecular substrates. Valuable insights into lysosome functions have emerged from research into these diseases. In addition to primary lysosomal dysfunction, cellular pathways associated with other membrane-bound organelles are perturbed in these disorders. Through selective examples, we illustrate why the term "cellular storage disorders" may be a more appropriate description of these diseases and discuss therapies that can alleviate storage and restore normal cellular function.
Figures



References
-
- Aqul A., Liu B., Ramirez C.M., Pieper A.A., Estill S.J., Burns D.K., Liu B., Repa J.J., Turley S.D., Dietschy J.M. 2011. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31:9404–9413 10.1523/JNEUROSCI.1317-11.2011 - DOI - PMC - PubMed
-
- Ballabio A. 2009. Disease pathogenesis explained by basic science: lysosomal storage diseases as autophagocytic disorders. Int. J. Clin. Pharmacol. Ther. 47(Suppl 1):S34–S38 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
