

Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells
- PMID: 23190711
- PMCID: PMC3526453
- DOI: 10.1186/1742-2094-9-260
Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells
Abstract
Background: Microglia, the resident macrophage-like cells in the brain, regulate innate immune responses in the CNS to protect neurons. However, excessive activation of microglia contributes to neurodegenerative diseases. Corticosteroids are potent modulators of inflammation and mediate their effects by binding to mineralocorticoid receptors (MR) and glucocorticoid receptors (GR). Here, the coordinated activities of GR and MR on the modulation of the nuclear factor-κB (NF-κB) pathway in murine BV-2 microglial cells were studied.
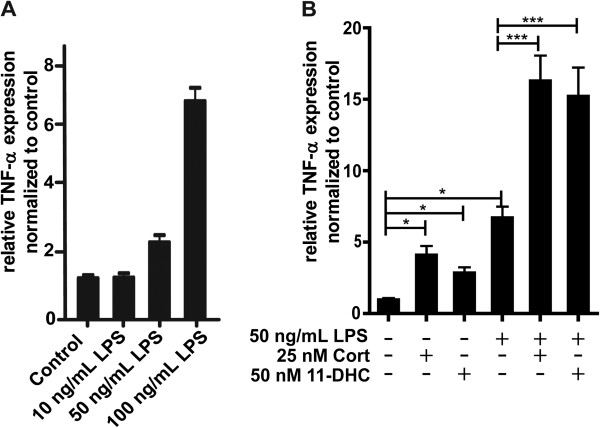
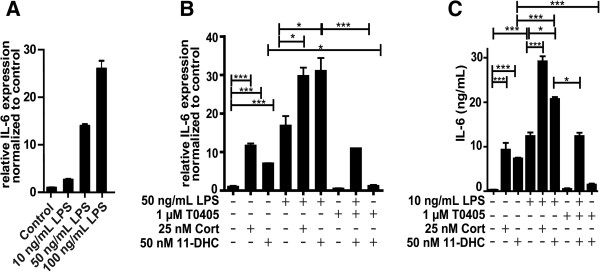
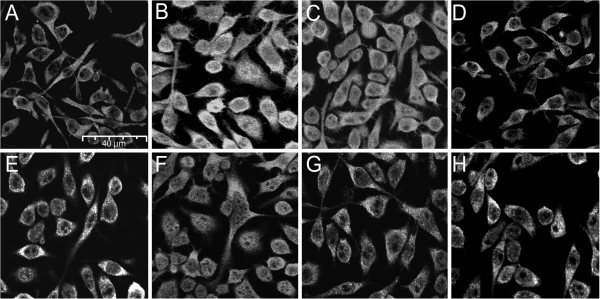
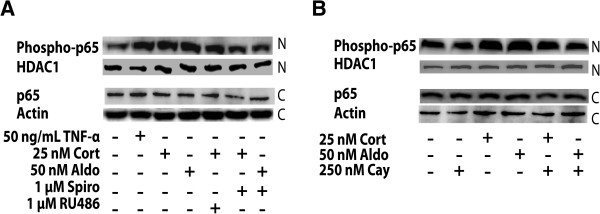
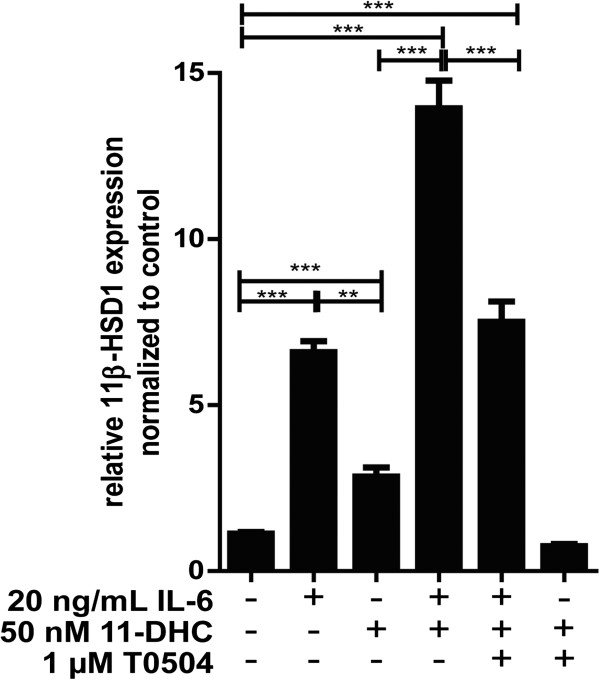
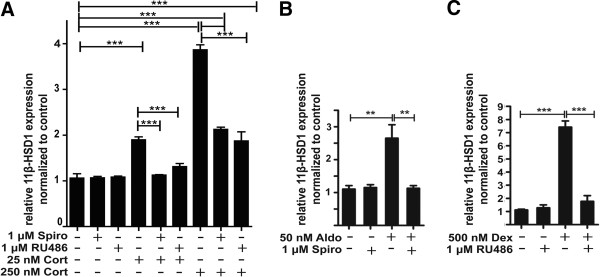
Methods: BV-2 cells were treated with different corticosteroids in the presence or absence of MR and GR antagonists. The impact of the glucocorticoid-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) was determined by incubating cells with 11-dehydrocorticosterone, with or without selective inhibitors. Expression of interleukin-6 (IL-6), tumor necrosis factor receptor 2 (TNFR2), and 11β-HSD1 mRNA was analyzed by RT-PCR and IL-6 protein expression by ELISA. NF-κB activation and translocation upon treatment with various corticosteroids were visualized by western blotting, immunofluorescence microscopy, and translocation assays.
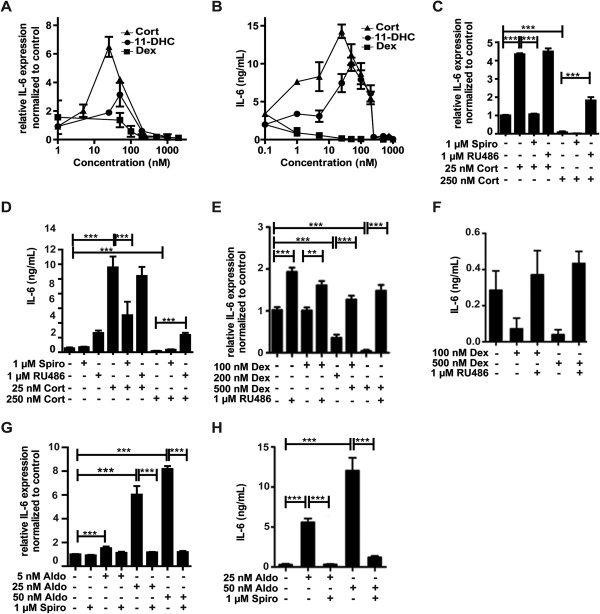
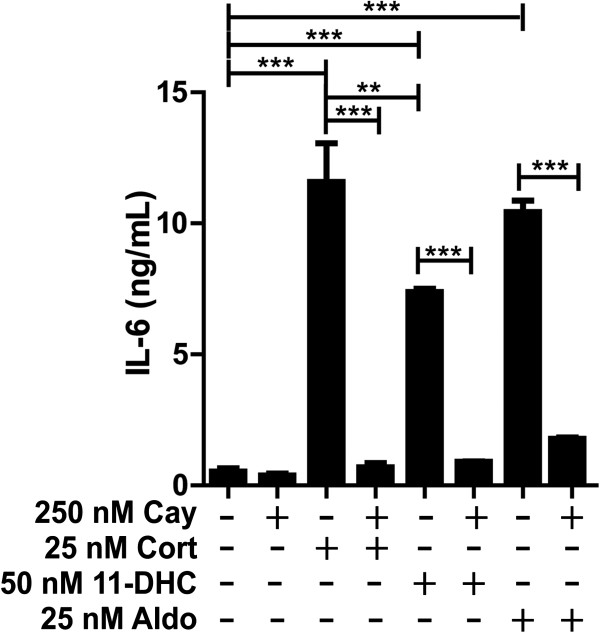
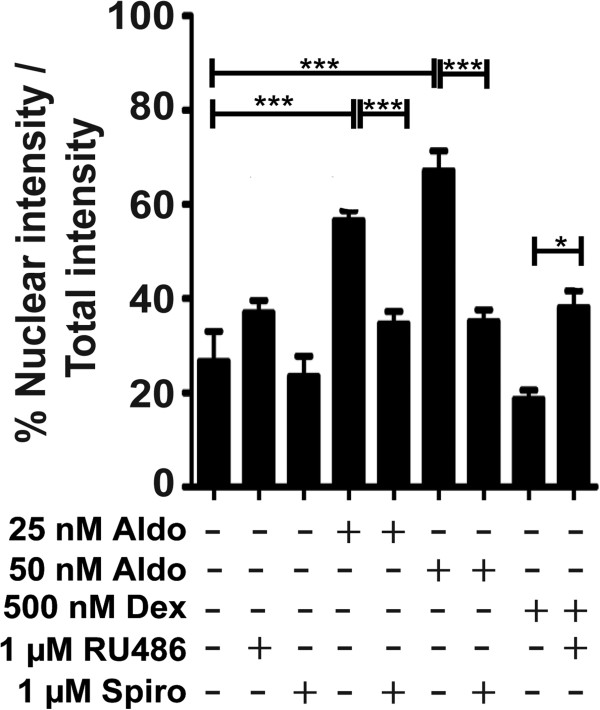
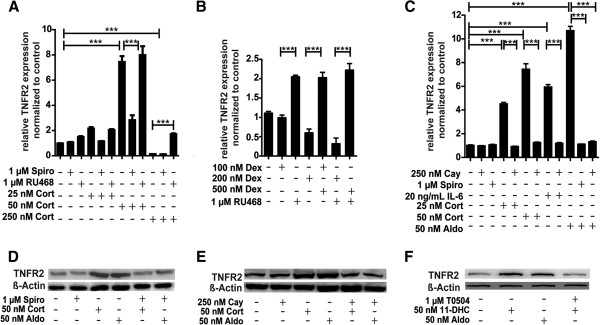
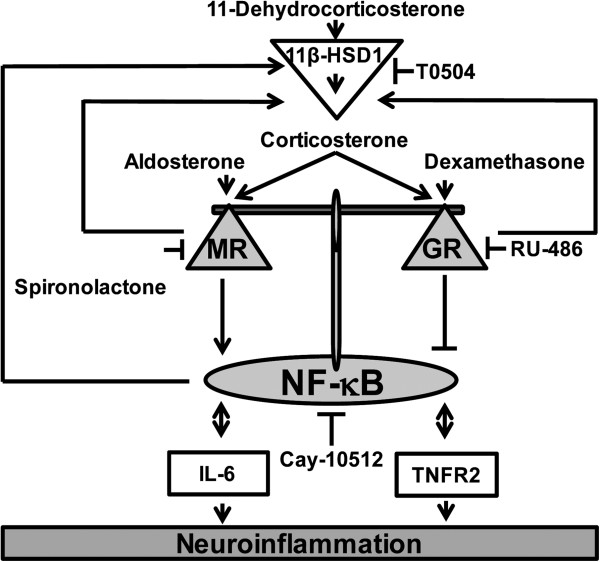
Results: GR and MR differentially regulate NF-κB activation and neuroinflammatory parameters in BV-2 cells. By converting inactive 11-dehydrocorticosterone to active corticosterone, 11β-HSD1 essentially modulates the coordinated action of GR and MR. Biphasic effects were observed for 11-dehydrocorticosterone and corticosterone, with an MR-dependent potentiation of IL-6 and tumor necrosis factor-α (TNF-α) expression and NF-κB activation at low/moderate concentrations and a GR-dependent suppression at high concentrations. The respective effects were confirmed using the MR ligand aldosterone and the antagonist spironolactone as well as the GR ligand dexamethasone and the antagonist RU-486. NF-κB activation could be blocked by spironolactone and the inhibitor of NF-κB translocation Cay-10512. Moreover, an increased expression of TNFR2 was observed upon treatment with 11-dehydrocorticosterone and aldosterone, which was reversed by 11β-HSD1 inhibitors and/or spironolactone and Cay-10512.
Conclusions: A tightly coordinated GR and MR activity regulates the NF-κB pathway and the control of inflammatory mediators in microglia cells. The balance of GR and MR activity is locally modulated by the action of 11β-HSD1, which is upregulated by pro-inflammatory mediators and may represent an important feedback mechanism involved in resolution of inflammation.
Figures
Similar articles
-
Toll-like receptor 4-dependent innate immune responses are mediated by intracrine corticosteroids and activation of glycogen synthase kinase-3β in astrocytes.FASEB J. 2024 Jul 15;38(13):e23781. doi: 10.1096/fj.202301923RR. FASEB J. 2024. PMID: 38941212
-
Decreased 11β-hydroxysteroid dehydrogenase 1 in lungs of steroid receptor coactivator (Src)-1/-2 double-deficient fetal mice is caused by impaired glucocorticoid and cytokine signaling.FASEB J. 2020 Dec;34(12):16243-16261. doi: 10.1096/fj.202001809R. Epub 2020 Oct 18. FASEB J. 2020. PMID: 33070362 Free PMC article.
-
11beta-hydroxysteroid dehydrogenase type 1 deficiency prevents memory deficits with aging by switching from glucocorticoid receptor to mineralocorticoid receptor-mediated cognitive control.J Neurosci. 2011 Mar 16;31(11):4188-93. doi: 10.1523/JNEUROSCI.6145-10.2011. J Neurosci. 2011. PMID: 21411659 Free PMC article.
-
Brain mineralocorticoid receptors in cognition and cardiovascular homeostasis.Steroids. 2014 Dec;91:20-31. doi: 10.1016/j.steroids.2014.08.014. Steroids. 2014. PMID: 25173821 Free PMC article. Review.
-
Changing glucocorticoid action: 11β-hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation.J Steroid Biochem Mol Biol. 2013 Sep;137:82-92. doi: 10.1016/j.jsbmb.2013.02.002. Epub 2013 Feb 19. J Steroid Biochem Mol Biol. 2013. PMID: 23435016 Free PMC article. Review.
Cited by
-
The Pro-inflammatory Effects of Glucocorticoids in the Brain.Front Endocrinol (Lausanne). 2016 Jun 28;7:78. doi: 10.3389/fendo.2016.00078. eCollection 2016. Front Endocrinol (Lausanne). 2016. PMID: 27445981 Free PMC article. Review.
-
Sugar-Modified Poly(propylene imine) Dendrimers Stimulate the NF-κB Pathway in a Myeloid Cell Line.Pharm Res. 2017 Jan;34(1):136-147. doi: 10.1007/s11095-016-2049-3. Epub 2016 Oct 20. Pharm Res. 2017. PMID: 27766462 Free PMC article.
-
Aldosterone exerts anti-inflammatory effects on LPS stimulated microglia.Heliyon. 2018 Oct 3;4(10):e00826. doi: 10.1016/j.heliyon.2018.e00826. eCollection 2018 Oct. Heliyon. 2018. PMID: 30302409 Free PMC article.
-
Enteric Microbiota⁻Gut⁻Brain Axis from the Perspective of Nuclear Receptors.Int J Mol Sci. 2018 Jul 28;19(8):2210. doi: 10.3390/ijms19082210. Int J Mol Sci. 2018. PMID: 30060580 Free PMC article. Review.
-
Corticosterone Preexposure Increases NF-κB Translocation and Sensitizes IL-1β Responses in BV2 Microglia-Like Cells.Front Immunol. 2018 Jan 22;9:3. doi: 10.3389/fimmu.2018.00003. eCollection 2018. Front Immunol. 2018. PMID: 29403490 Free PMC article.
References
-
- Ishii-Yonemoto T, Masuzaki H, Yasue S, Okada S, Kozuka C, Tanaka T, Noguchi M, Tomita T, Fujikura J, Yamamoto Y, Ebihara K, Hosoda K, Nakao K. Glucocorticoid reamplification within cells intensifies NF-kappaB and MAPK signaling and reinforces inflammation in activated preadipocytes. Am J Physiol Endocrinol Metab. 2010;298:E930–E940. doi: 10.1152/ajpendo.00320.2009. - DOI - PubMed
-
- De Kloet ER, Veldhuis HD, Wagenaars JL, Bergink EW. Relative binding affinity of steroids for the corticosterone receptor system in rat hippocampus. J Steroid Biochem Mol Biol. 1984;21:173–178. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
