

Protein kinase C, an elusive therapeutic target?
- PMID: 23197040
- PMCID: PMC3760692
- DOI: 10.1038/nrd3871
Protein kinase C, an elusive therapeutic target?
Abstract
Protein kinase C (PKC) has been a tantalizing target for drug discovery ever since it was first identified as the receptor for the tumour promoter phorbol ester in 1982. Although initial therapeutic efforts focused on cancer, additional indications--including diabetic complications, heart failure, myocardial infarction, pain and bipolar disorder--were targeted as researchers developed a better understanding of the roles of eight conventional and novel PKC isozymes in health and disease. Unfortunately, both academic and pharmaceutical efforts have yet to result in the approval of a single new drug that specifically targets PKC. Why does PKC remain an elusive drug target? This Review provides a short account of some of the efforts, challenges and opportunities in developing PKC modulators to address unmet clinical needs.
Figures
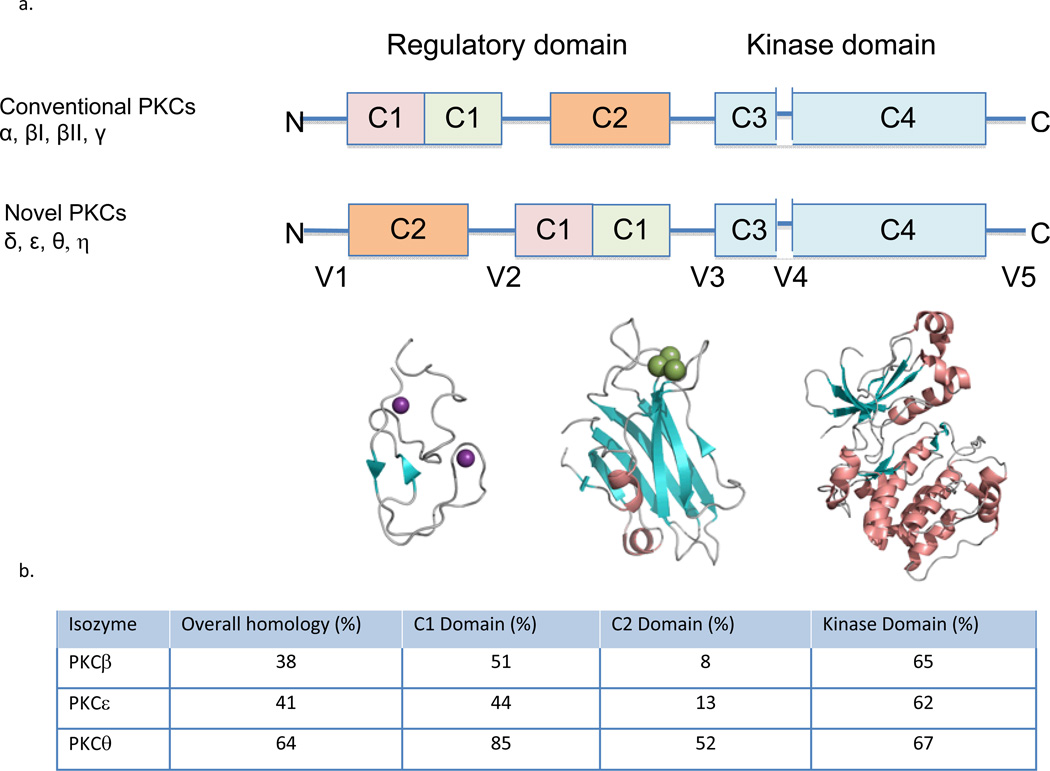
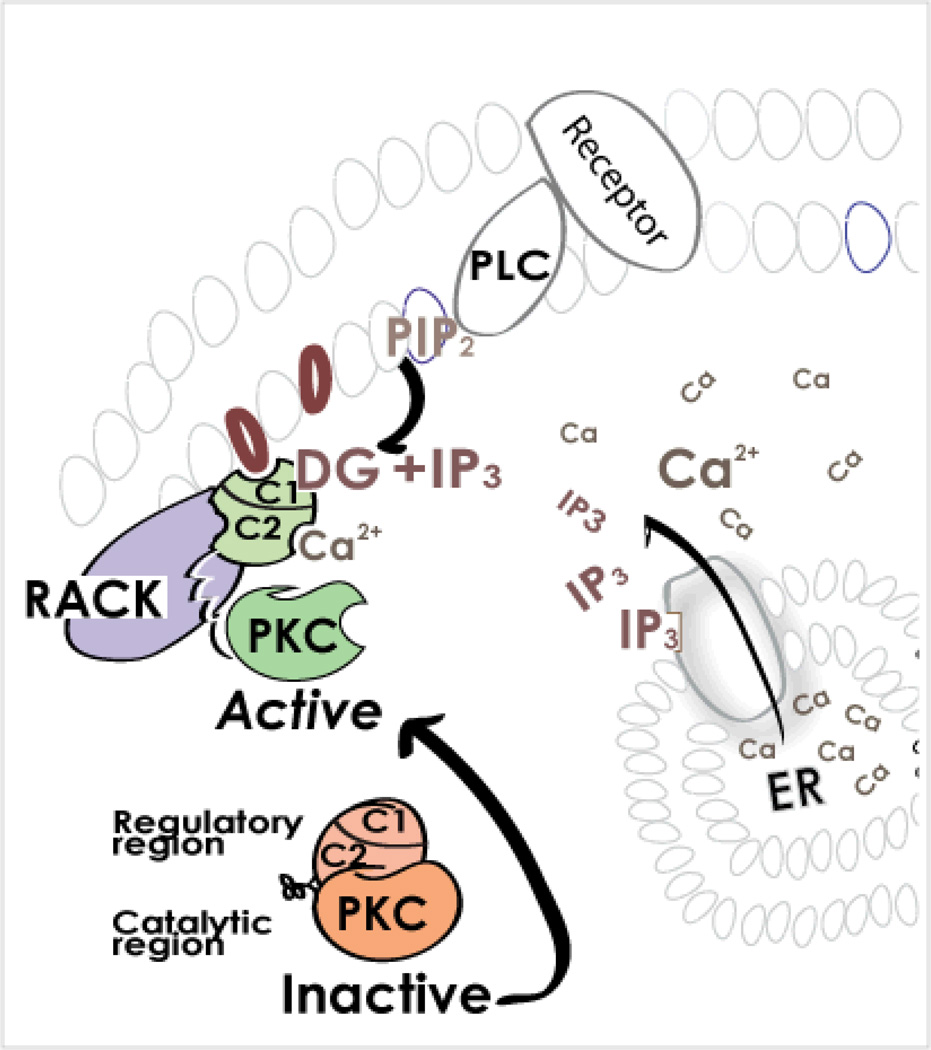
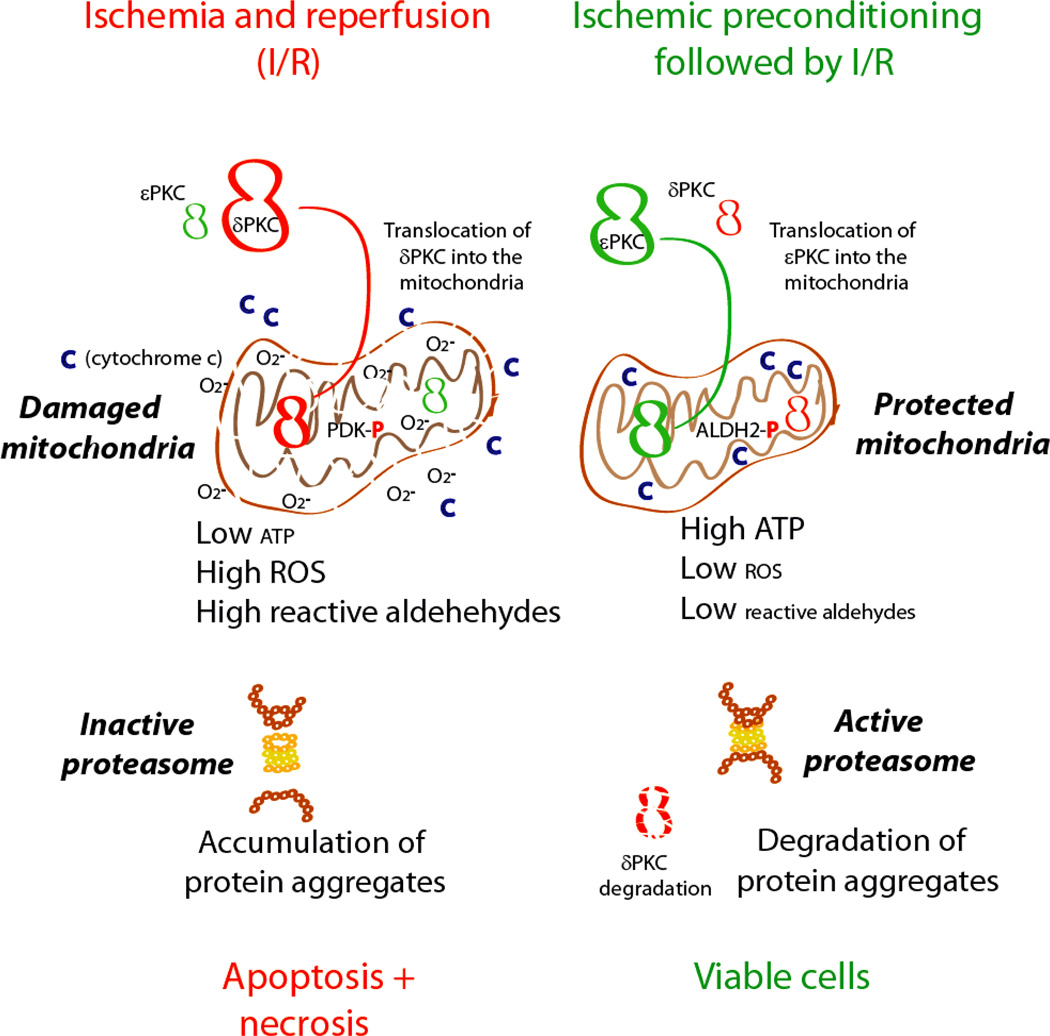
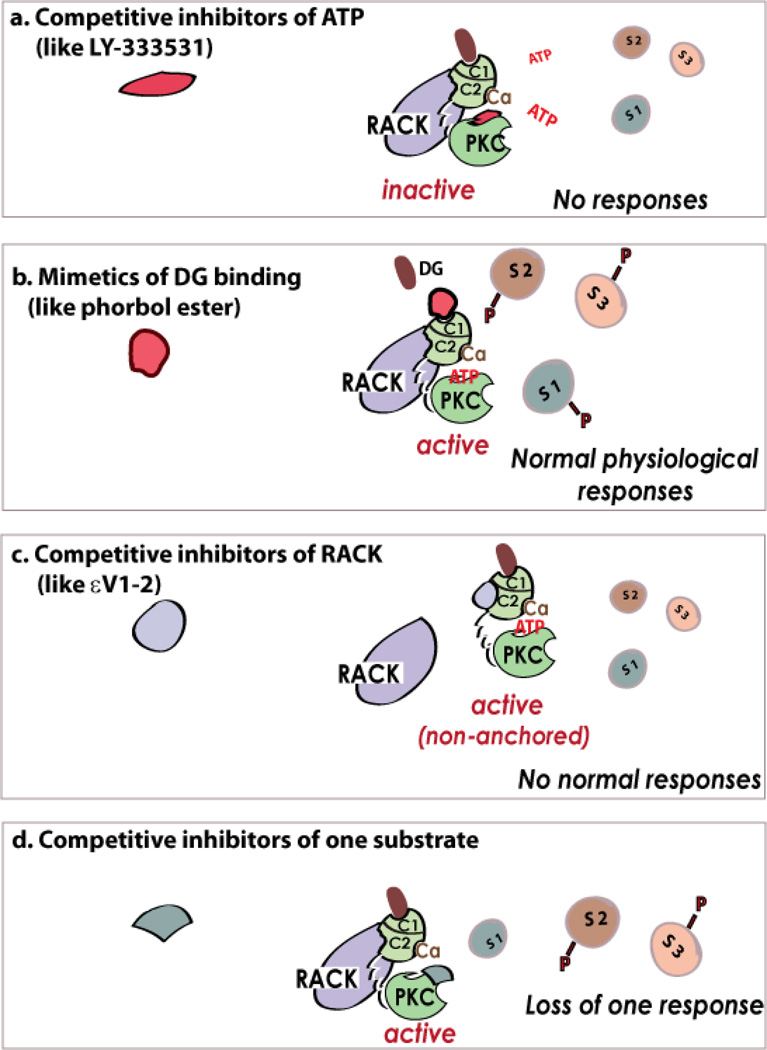
References
-
- Castagna M, et al. Direct activation of calcium-activated, phospholipid-dependent protein-kinase by tumor-promoting phorbo esters. J Biol Chem. 1982;257:7847–7851. - PubMed
-
- Toton E, Ignatowicz E, Skrzeczkowska K, Rybczynska M. Protein kinase Cepsilon as a cancer marker and target for anticancer therapy. Pharmacol Rep. 2011;63:19–29. - PubMed
-
- Inagaki K, Churchill E, Mochly-Rosen D. Epsilon protein kinase C as a potential therapeutic target for the ischemic heart. Cardiovasc Res. 2006;70:222–230. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
