

Amyotrophic lateral sclerosis model derived from human embryonic stem cells overexpressing mutant superoxide dismutase 1
- PMID: 23197818
- PMCID: PMC3659703
- DOI: 10.5966/sctm.2011-0061
Amyotrophic lateral sclerosis model derived from human embryonic stem cells overexpressing mutant superoxide dismutase 1
Abstract
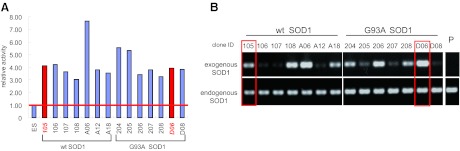
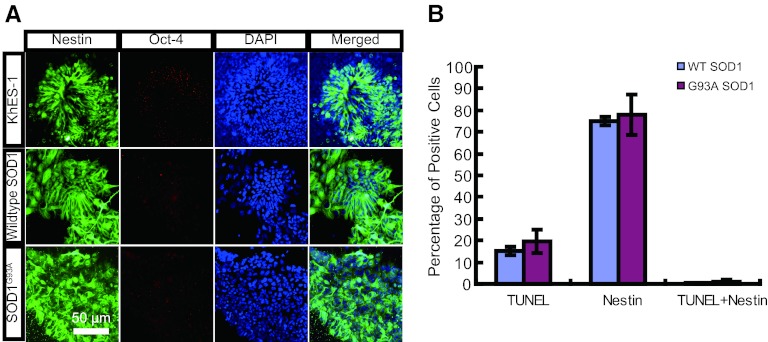
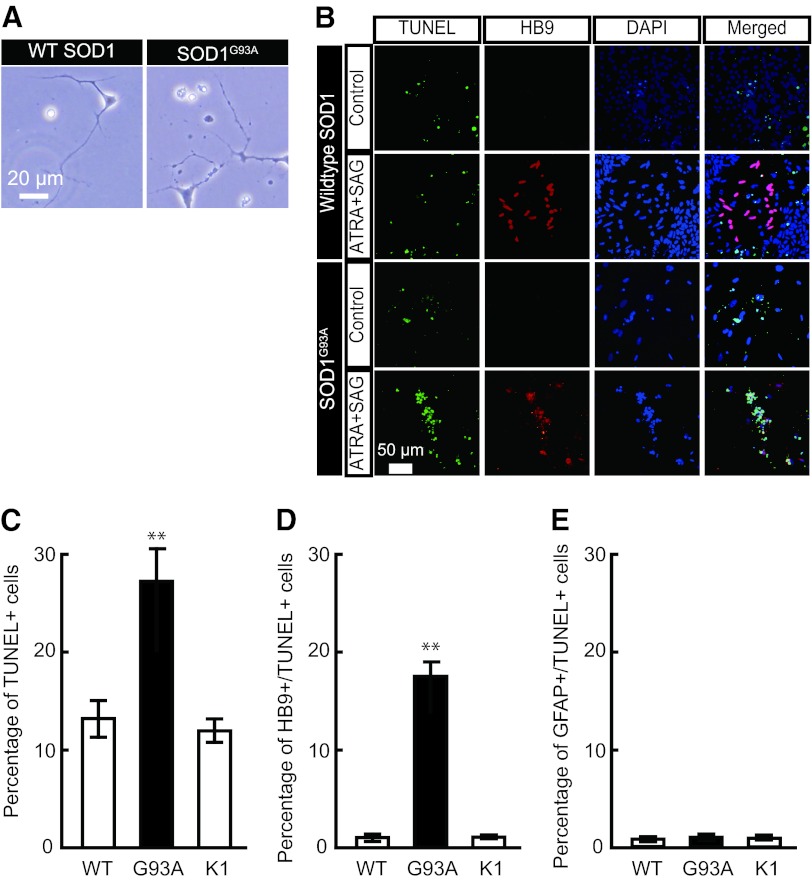
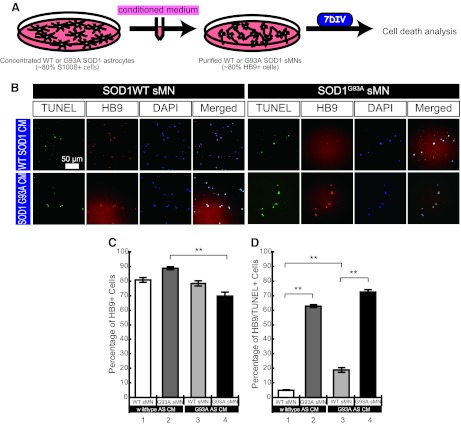
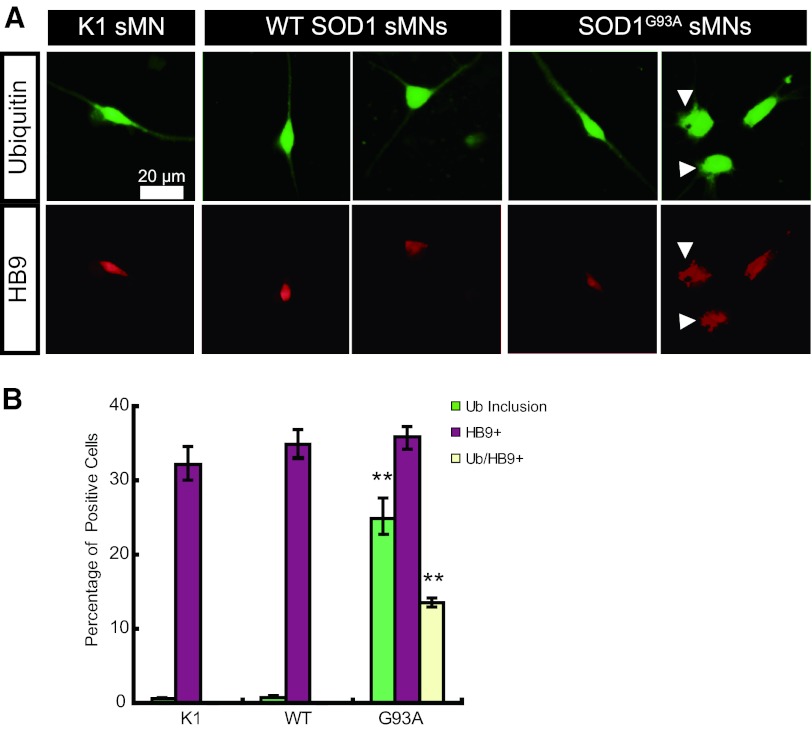
The generation of amyotrophic lateral sclerosis (ALS) disease models is an important subject for investigating disease mechanisms and pharmaceutical applications. In transgenic mice, expression of a mutant form of superoxide dismutase 1 (SOD1) can lead to the development of ALS that closely mimics the familial type of ALS (FALS). Although SOD1 mutant mice show phenotypes similar to FALS, dissimilar drug responses and size differences limit their usefulness to study the disease mechanism(s) and identify potential therapeutic compounds. Development of an in vitro model system for ALS is expected to help in obtaining novel insights into disease mechanisms and discovery of therapeutics. We report the establishment of an in vitro FALS model from human embryonic stem cells overexpressing either a wild-type (WT) or a mutant SOD1 (G93A) gene and the evaluation of the phenotypes and survival of the spinal motor neurons (sMNs), which are the neurons affected in ALS patients. The in vitro FALS model that we developed mimics the in vivo human ALS disease in terms of the following: (a) selective degeneration of sMNs expressing the G93A SOD1 but not those expressing the WT gene; (b) susceptibility of G93A SOD1-derived sMNs to form ubiquitinated inclusions; (c) astrocyte-derived factor(s) in the selective degeneration of G93A SOD1 sMNs; and (d) cell-autonomous, as well as non-cell-autonomous, dependent sMN degeneration. Thus, this model is expected to help unravel the disease mechanisms involved in the development of FALS and also lead to potential drug discoveries based on the prevention of neurodegeneration.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
