

Tips and tricks for the assembly of a Corynebacterium pseudotuberculosis genome using a semiconductor sequencer
- PMID: 23199210
- PMCID: PMC3917457
- DOI: 10.1111/1751-7915.12006
Tips and tricks for the assembly of a Corynebacterium pseudotuberculosis genome using a semiconductor sequencer
Abstract
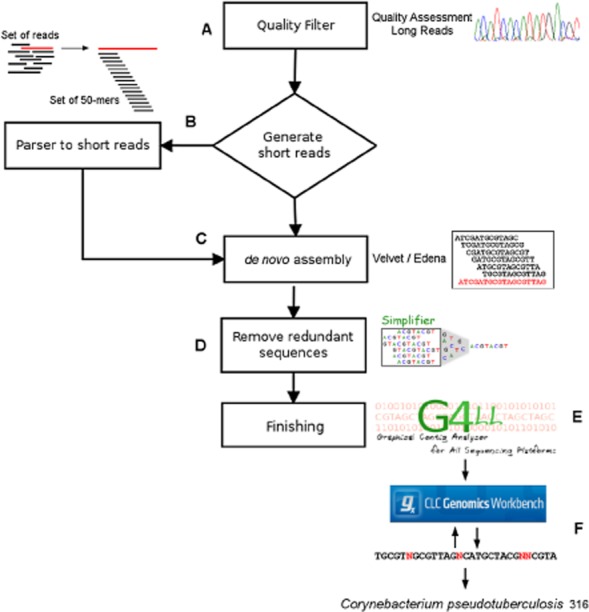
New sequencing platforms have enabled rapid decoding of complete prokaryotic genomes at relatively low cost. The Ion Torrent platform is an example of these technologies, characterized by lower coverage, generating challenges for the genome assembly. One particular problem is the lack of genomes that enable reference-based assembly, such as the one used in the present study, Corynebacterium pseudotuberculosis biovar equi, which causes high economic losses in the US equine industry. The quality treatment strategy incorporated into the assembly pipeline enabled a 16-fold greater use of the sequencing data obtained compared with traditional quality filter approaches. Data preprocessing prior to the de novo assembly enabled the use of known methodologies in the next-generation sequencing data assembly. Moreover, manual curation was proved to be essential for ensuring a quality assembly, which was validated by comparative genomics with other species of the genus Corynebacterium. The present study presents a modus operandi that enables a greater and better use of data obtained from semiconductor sequencing for obtaining the complete genome from a prokaryotic microorganism, C. pseudotuberculosis, which is not a traditional biological model such as Escherichia coli.
© 2012 The Authors. Published by Society for Applied Microbiology and Blackwell Publishing Ltd. This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Figures
Similar articles
-
Genome sequence of the Corynebacterium pseudotuberculosis Cp316 strain, isolated from the abscess of a Californian horse.J Bacteriol. 2012 Dec;194(23):6620-1. doi: 10.1128/JB.01616-12. J Bacteriol. 2012. PMID: 23144380 Free PMC article.
-
Assessing the Genotypic Differences between Strains of Corynebacterium pseudotuberculosis biovar equi through Comparative Genomics.PLoS One. 2017 Jan 26;12(1):e0170676. doi: 10.1371/journal.pone.0170676. eCollection 2017. PLoS One. 2017. PMID: 28125655 Free PMC article.
-
Complete genome sequence of Corynebacterium pseudotuberculosis strain CIP 52.97, isolated from a horse in Kenya.J Bacteriol. 2011 Dec;193(24):7025-6. doi: 10.1128/JB.06293-11. J Bacteriol. 2011. PMID: 22123771 Free PMC article.
-
Brazilian genome sequencing projects: state of the art.Recent Pat DNA Gene Seq. 2008;2(2):111-32. doi: 10.2174/187221508784534203. Recent Pat DNA Gene Seq. 2008. PMID: 19075951 Review.
-
Progress in ion torrent semiconductor chip based sequencing.Electrophoresis. 2012 Dec;33(23):3397-417. doi: 10.1002/elps.201200424. Electrophoresis. 2012. PMID: 23208921 Review.
Cited by
-
Draft Genome Sequence of Non-O1 and Non-O139 Vibrio cholerae Strain VCC19.Genome Announc. 2014 Nov 6;2(6):e01094-14. doi: 10.1128/genomeA.01094-14. Genome Announc. 2014. PMID: 25377699 Free PMC article.
-
Exploration of Nitrate Reductase Metabolic Pathway in Corynebacterium pseudotuberculosis.Int J Genomics. 2017;2017:9481756. doi: 10.1155/2017/9481756. Epub 2017 Feb 20. Int J Genomics. 2017. PMID: 28316974 Free PMC article.
-
Genomic analysis of four strains of Corynebacterium pseudotuberculosis bv. Equi isolated from horses showing distinct signs of infection.Stand Genomic Sci. 2017 Jan 31;12:16. doi: 10.1186/s40793-017-0234-6. eCollection 2017. Stand Genomic Sci. 2017. PMID: 28163825 Free PMC article.
-
Comparative genomic analysis between Corynebacterium pseudotuberculosis strains isolated from buffalo.PLoS One. 2017 Apr 26;12(4):e0176347. doi: 10.1371/journal.pone.0176347. eCollection 2017. PLoS One. 2017. PMID: 28445543 Free PMC article.
-
The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains.PLoS One. 2013;8(1):e53818. doi: 10.1371/journal.pone.0053818. Epub 2013 Jan 14. PLoS One. 2013. PMID: 23342011 Free PMC article.
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. - PubMed
-
- Cerdeira LT, Carneiro AR, Ramos RTJ, de Almeida SS, D'Afonseca V, Schneider MPC, et al. Rapid hybrid de novo assembly of a microbial genome using only short reads: Corynebacterium pseudotuberculosis I19 as a case study. J Microbiol Methods. 2011;86:218–223. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
