

Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior
- PMID: 23201971
- PMCID: PMC3979286
- DOI: 10.1038/nn.3277
Evolution of GluN2A/B cytoplasmic domains diversified vertebrate synaptic plasticity and behavior
Erratum in
- Nat Neurosci. 2013 Dec;16(12):1908
Abstract
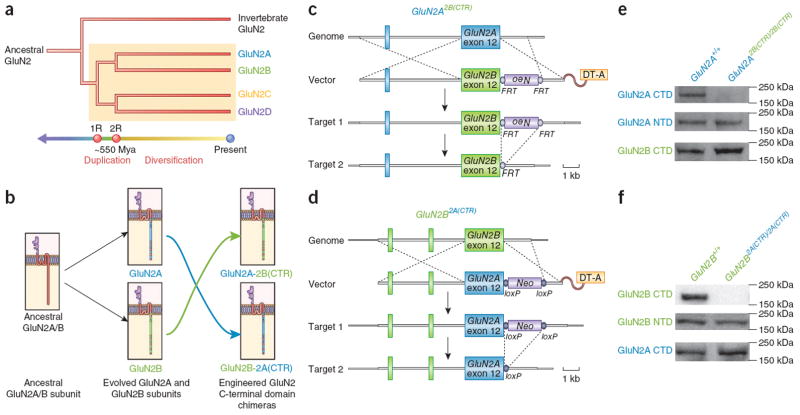
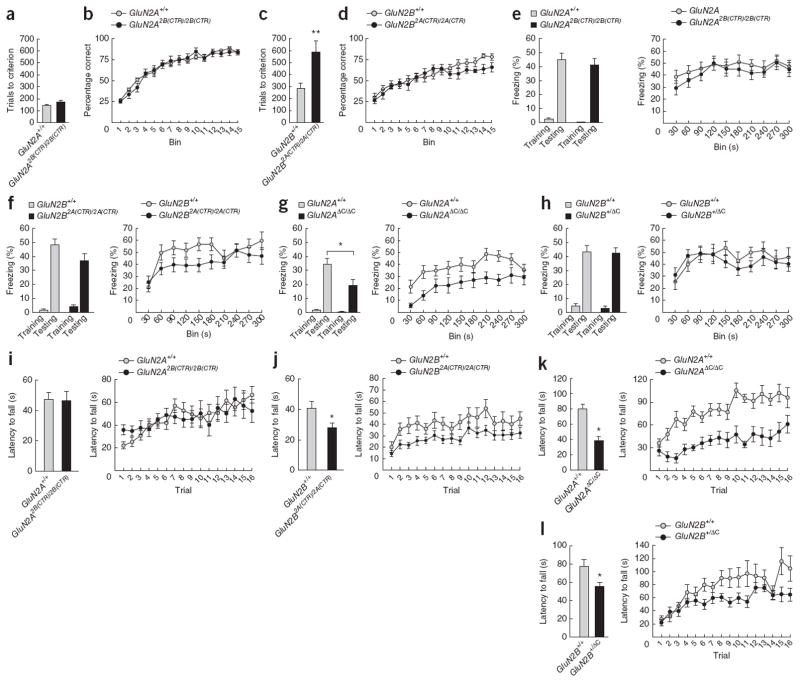
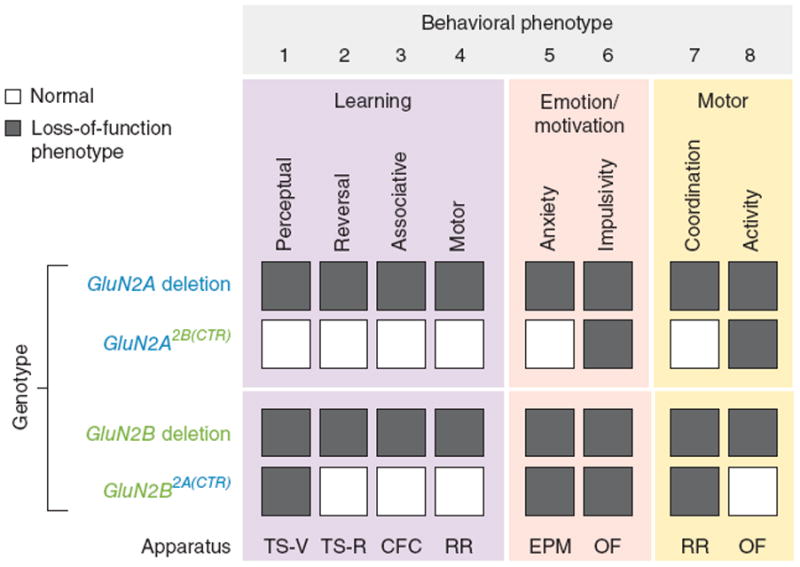
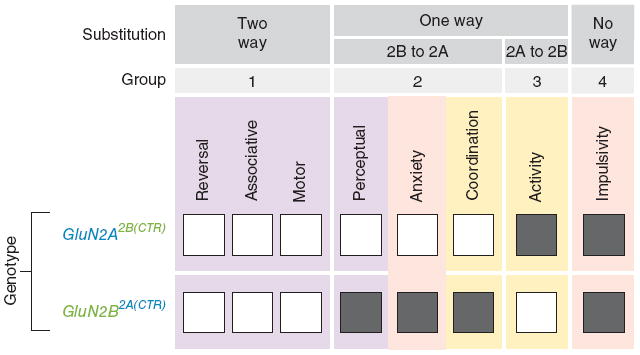
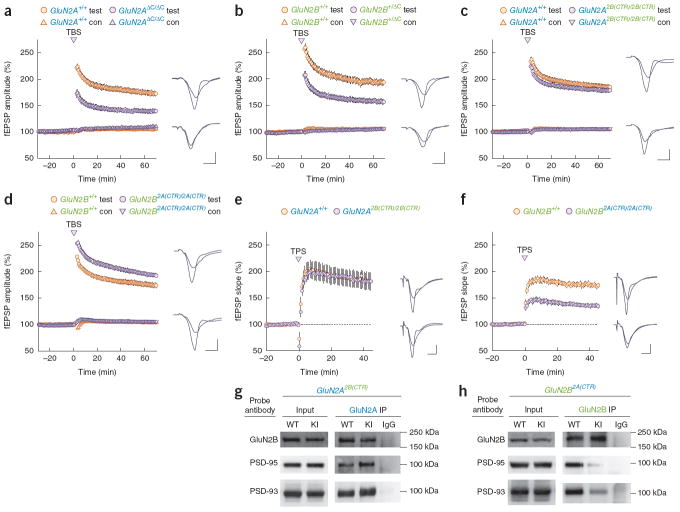
Two genome duplications early in the vertebrate lineage expanded gene families, including GluN2 subunits of the NMDA receptor. Diversification between the four mammalian GluN2 proteins occurred primarily at their intracellular C-terminal domains (CTDs). To identify shared ancestral functions and diversified subunit-specific functions, we exchanged the exons encoding the GluN2A (also known as Grin2a) and GluN2B (also known as Grin2b) CTDs in two knock-in mice and analyzed the mice's biochemistry, synaptic physiology, and multiple learned and innate behaviors. The eight behaviors were genetically separated into four groups, including one group comprising three types of learning linked to conserved GluN2A/B regions. In contrast, the remaining five behaviors exhibited subunit-specific regulation. GluN2A/B CTD diversification conferred differential binding to cytoplasmic MAGUK proteins and differential forms of long-term potentiation. These data indicate that vertebrate behavior and synaptic signaling acquired increased complexity from the duplication and diversification of ancestral GluN2 genes.
Figures


Comment in
-
Retooling spare parts: gene duplication and cognition.Nat Neurosci. 2013 Jan;16(1):6-8. doi: 10.1038/nn.3292. Nat Neurosci. 2013. PMID: 23257927 Free PMC article.
References
-
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. - PubMed
-
- LeDoux JE. Synaptic Self: How Our Brains Become Who We Are. Viking Press; 2002.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
