

dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection
- PMID: 23202496
- PMCID: PMC3509664
- DOI: 10.3390/v4112598
dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection
Abstract
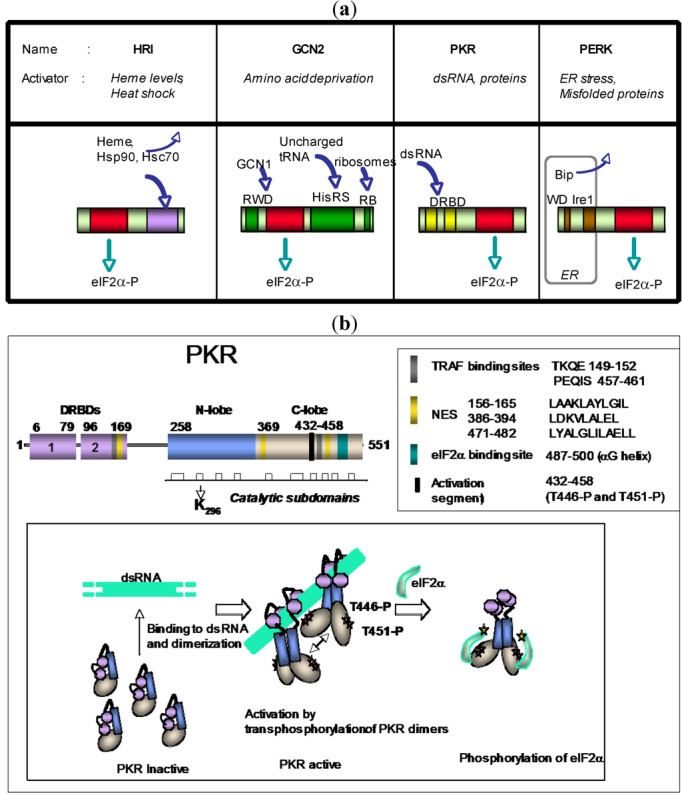
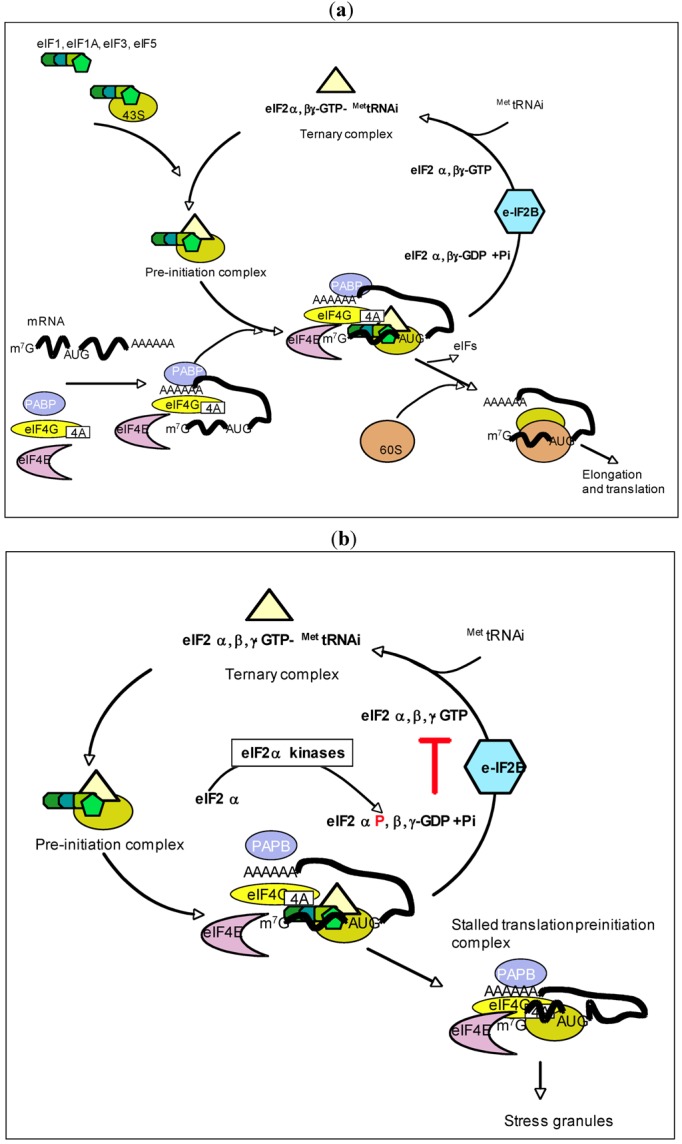
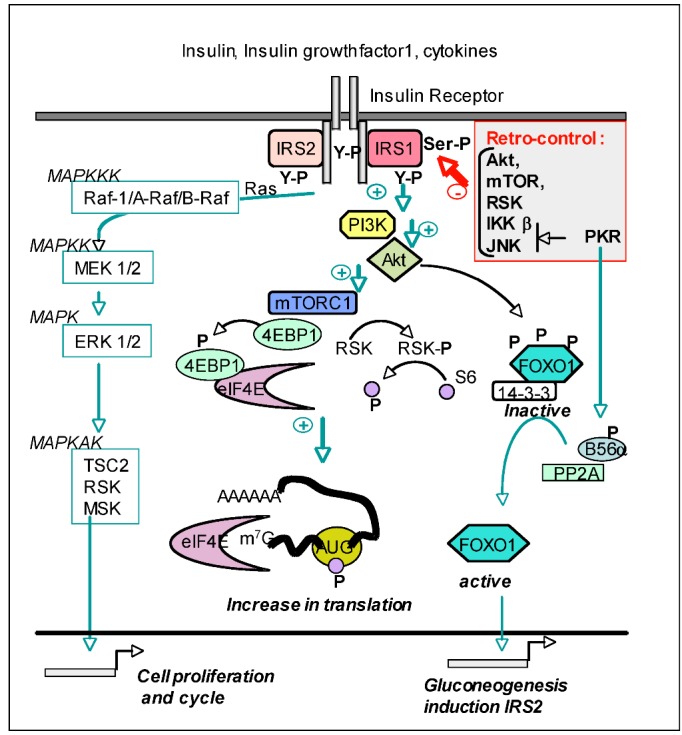
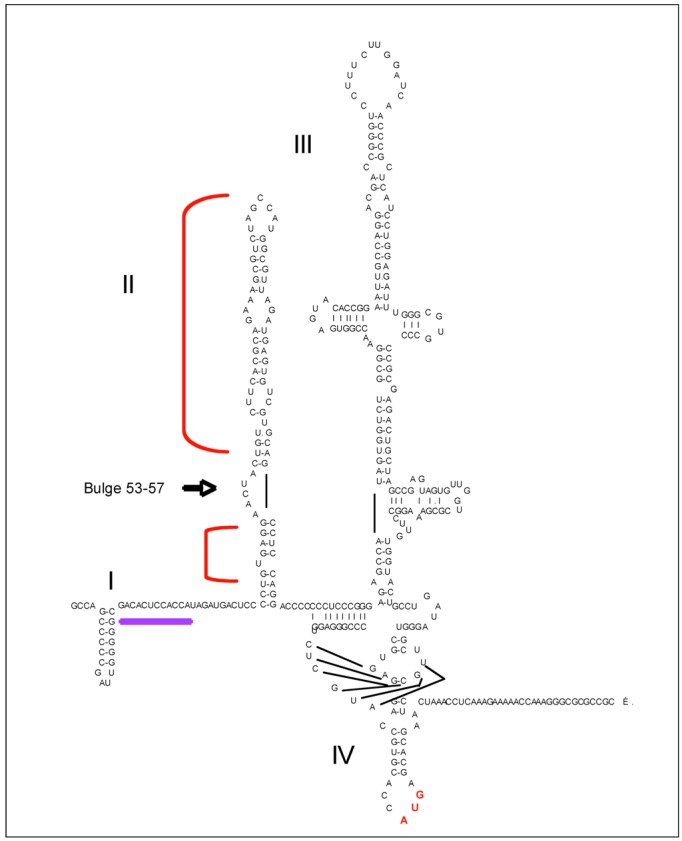
The double-stranded RNA-dependent protein kinase PKR plays multiple roles in cells, in response to different stress situations. As a member of the interferon (IFN)‑Stimulated Genes, PKR was initially recognized as an actor in the antiviral action of IFN, due to its ability to control translation, through phosphorylation, of the alpha subunit of eukaryotic initiation factor 2 (eIF2a). As such, PKR participates in the generation of stress granules, or autophagy and a number of viruses have designed strategies to inhibit its action. However, PKR deficient mice resist most viral infections, indicating that PKR may play other roles in the cell other than just acting as an antiviral agent. Indeed, PKR regulates several signaling pathways, either as an adapter protein and/or using its kinase activity. Here we review the role of PKR as an eIF2a kinase, its participation in the regulation of the NF-kB, p38MAPK and insulin pathways, and we focus on its role during infection with the hepatitis C virus (HCV). PKR binds the HCV IRES RNA, cooperates with some functions of the HCV core protein and may represent a target for NS5A or E2. Novel data points out for a role of PKR as a pro-HCV agent, both as an adapter protein and as an eIF2a-kinase, and in cooperation with the di-ubiquitin-like protein ISG15. Developing pharmaceutical inhibitors of PKR may help in resolving some viral infections as well as stress-related damages.
Figures
Similar articles
-
Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase.J Virol. 1999 Aug;73(8):6506-16. doi: 10.1128/JVI.73.8.6506-6516.1999. J Virol. 1999. PMID: 10400746 Free PMC article.
-
Hepatitis C virus controls interferon production through PKR activation.PLoS One. 2010 May 11;5(5):e10575. doi: 10.1371/journal.pone.0010575. PLoS One. 2010. PMID: 20485506 Free PMC article.
-
HCV NS5A co-operates with PKR in modulating HCV IRES-dependent translation.Infect Genet Evol. 2014 Aug;26:113-22. doi: 10.1016/j.meegid.2014.04.015. Epub 2014 May 9. Infect Genet Evol. 2014. PMID: 24815730
-
Roles of protein kinase R in cancer: Potential as a therapeutic target.Cancer Sci. 2018 Apr;109(4):919-925. doi: 10.1111/cas.13551. Epub 2018 Mar 23. Cancer Sci. 2018. PMID: 29478262 Free PMC article. Review.
-
PKR--a protein kinase regulated by double-stranded RNA.Int J Biochem Cell Biol. 1997 Jul;29(7):945-9. doi: 10.1016/s1357-2725(96)00169-0. Int J Biochem Cell Biol. 1997. PMID: 9375375 Review.
Cited by
-
Hepatitis C virus comes for dinner: How the hepatitis C virus interferes with autophagy.World J Gastroenterol. 2015 Jul 28;21(28):8492-507. doi: 10.3748/wjg.v21.i28.8492. World J Gastroenterol. 2015. PMID: 26229393 Free PMC article. Review.
-
Inhibition of mitochondrial complex III or dihydroorotate dehydrogenase (DHODH) triggers formation of poly(A)+ RNA foci adjacent to nuclear speckles following activation of ATM (ataxia telangiectasia mutated).RNA Biol. 2022 Jan;19(1):1244-1255. doi: 10.1080/15476286.2022.2146919. RNA Biol. 2022. PMID: 36412986 Free PMC article.
-
Enhanced Replication of Mouse Adenovirus Type 1 following Virus-Induced Degradation of Protein Kinase R (PKR).mBio. 2019 Apr 23;10(2):e00668-19. doi: 10.1128/mBio.00668-19. mBio. 2019. PMID: 31015330 Free PMC article.
-
A brave new world of RNA-binding proteins.Nat Rev Mol Cell Biol. 2018 May;19(5):327-341. doi: 10.1038/nrm.2017.130. Epub 2018 Jan 17. Nat Rev Mol Cell Biol. 2018. PMID: 29339797 Review.
-
2-aminopurine suppresses the TGF-β1-induced epithelial-mesenchymal transition and attenuates bleomycin-induced pulmonary fibrosis.Cell Death Discov. 2018 Feb 13;4:17. doi: 10.1038/s41420-017-0016-3. eCollection 2018 Dec. Cell Death Discov. 2018. PMID: 29531814 Free PMC article.
References
-
- Saunders L.R., Barber G.N. The dsRNA binding protein family: Critical roles, diverse cellular functions. FASEB J. 2003;17:961–983. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
