

Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes
- PMID: 23202730
- PMCID: PMC3533290
- DOI: 10.1172/JCI64516
Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes
Abstract
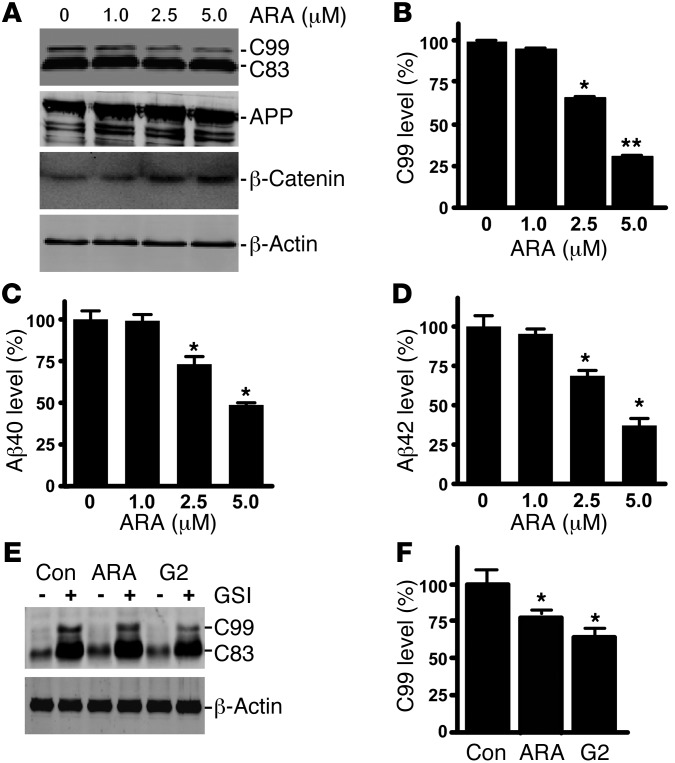
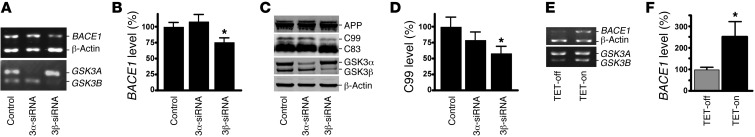
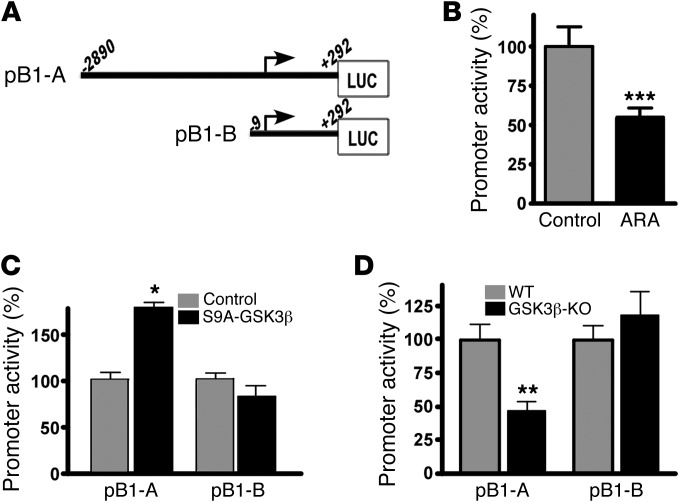
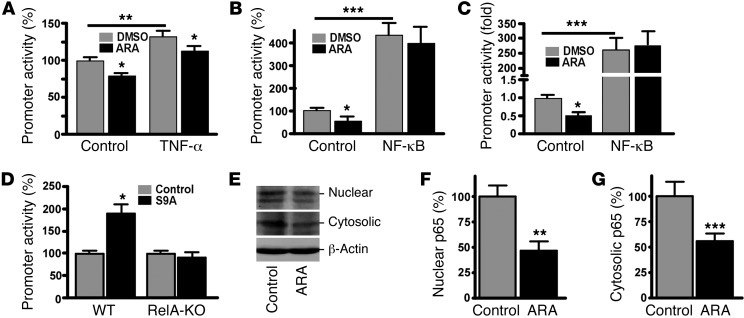
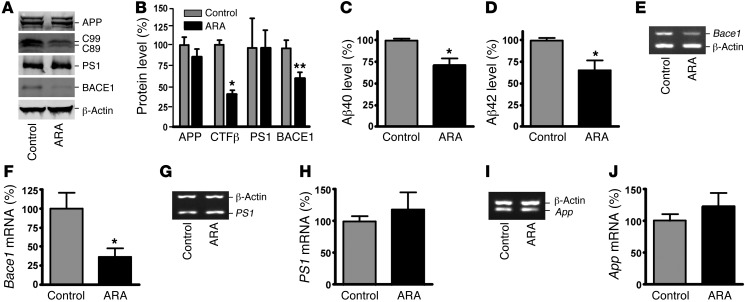
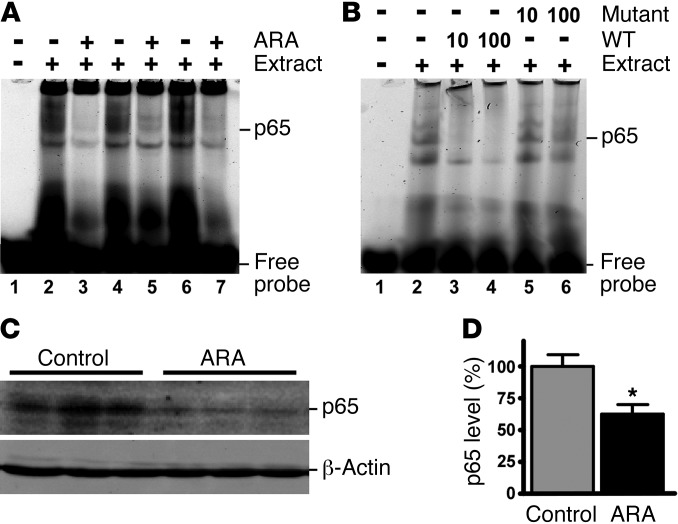
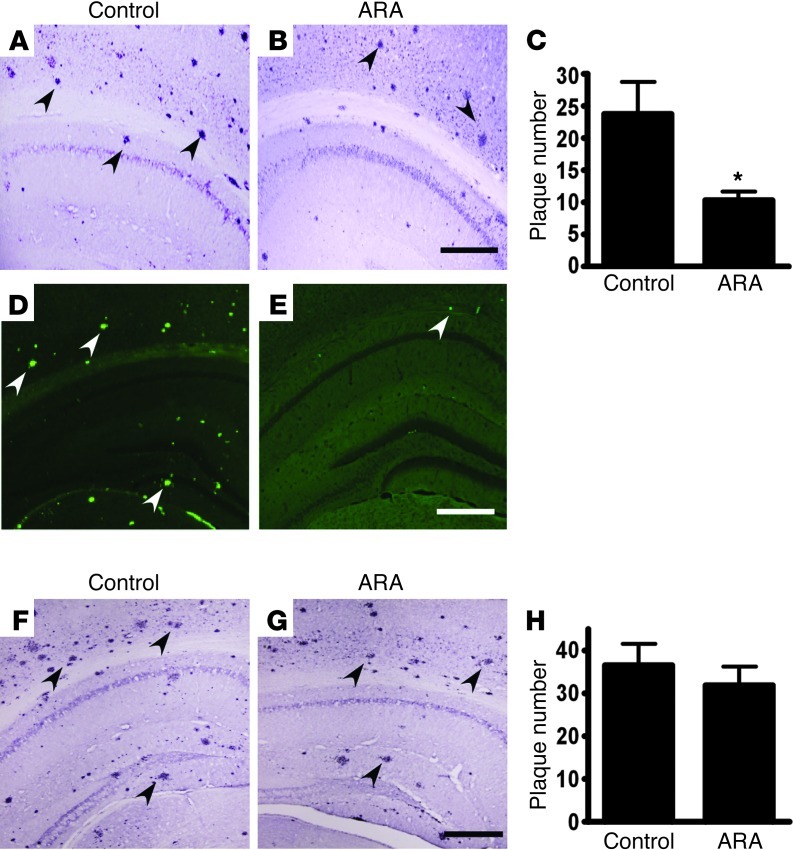
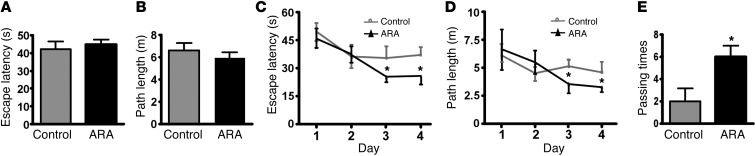
Deposition of amyloid β protein (Aβ) to form neuritic plaques in the brain is the pathological hallmark of Alzheimer's disease (AD). Aβ is generated from sequential cleavages of the β-amyloid precursor protein (APP) by the β- and γ-secretases, and β-site APP-cleaving enzyme 1 (BACE1) is the β-secretase essential for Aβ generation. Previous studies have indicated that glycogen synthase kinase 3 (GSK3) may play a role in APP processing by modulating γ-secretase activity, thereby facilitating Aβ production. There are two highly conserved isoforms of GSK3: GSK3α and GSK3β. We now report that specific inhibition of GSK3β, but not GSK3α, reduced BACE1-mediated cleavage of APP and Aβ production by decreasing BACE1 gene transcription and expression. The regulation of BACE1 gene expression by GSK3β was dependent on NF-κB signaling. Inhibition of GSK3 signaling markedly reduced Aβ deposition and neuritic plaque formation, and rescued memory deficits in the double transgenic AD model mice. These data provide evidence for regulation of BACE1 expression and AD pathogenesis by GSK3β and that inhibition of GSK3 signaling can reduce Aβ neuropathology and alleviate memory deficits in AD model mice. Our study suggests that interventions that specifically target the β-isoform of GSK3 may be a safe and effective approach for treating AD.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
