

Dfam: a database of repetitive DNA based on profile hidden Markov models
- PMID: 23203985
- PMCID: PMC3531169
- DOI: 10.1093/nar/gks1265
Dfam: a database of repetitive DNA based on profile hidden Markov models
Abstract
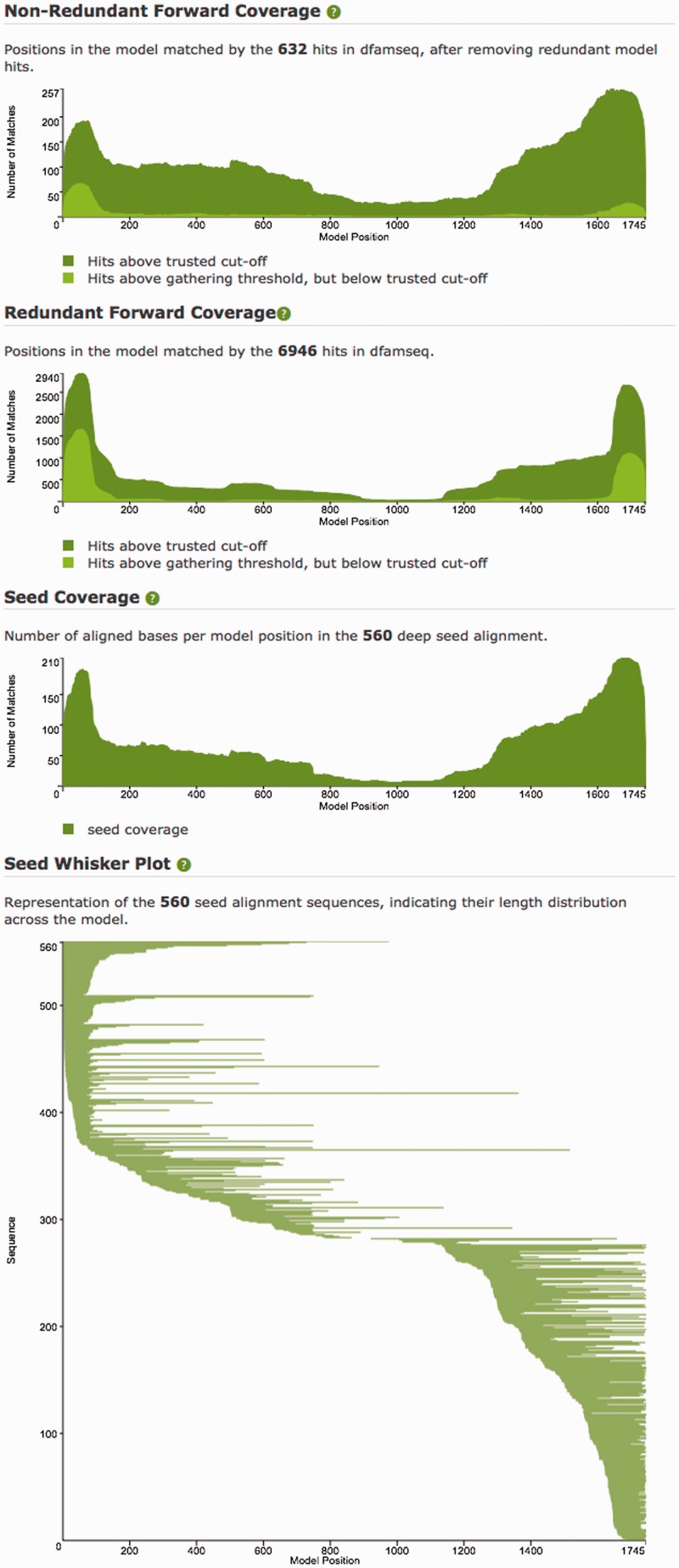
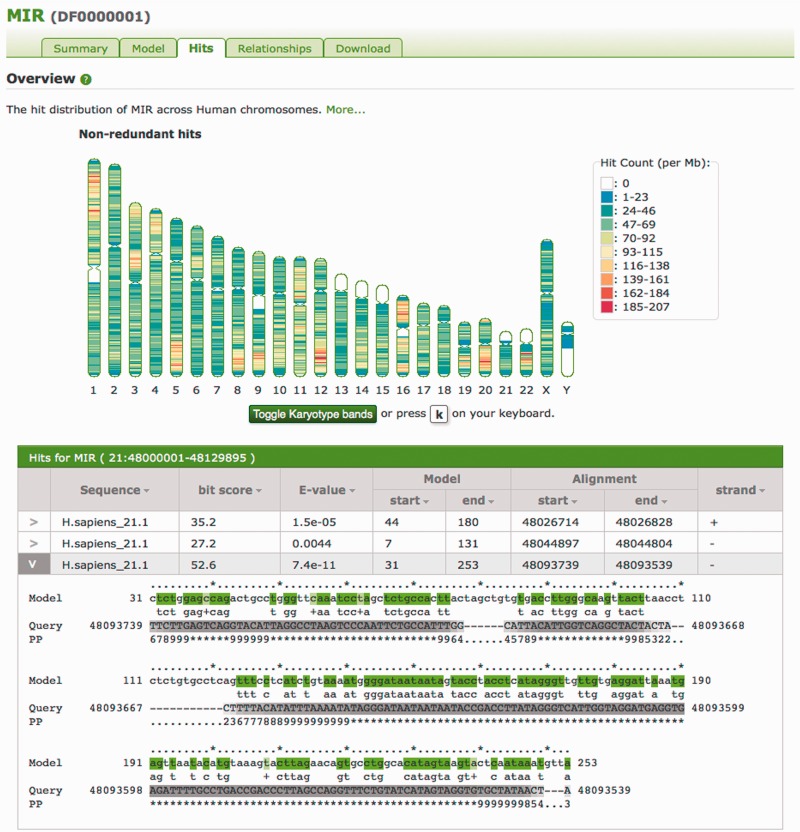
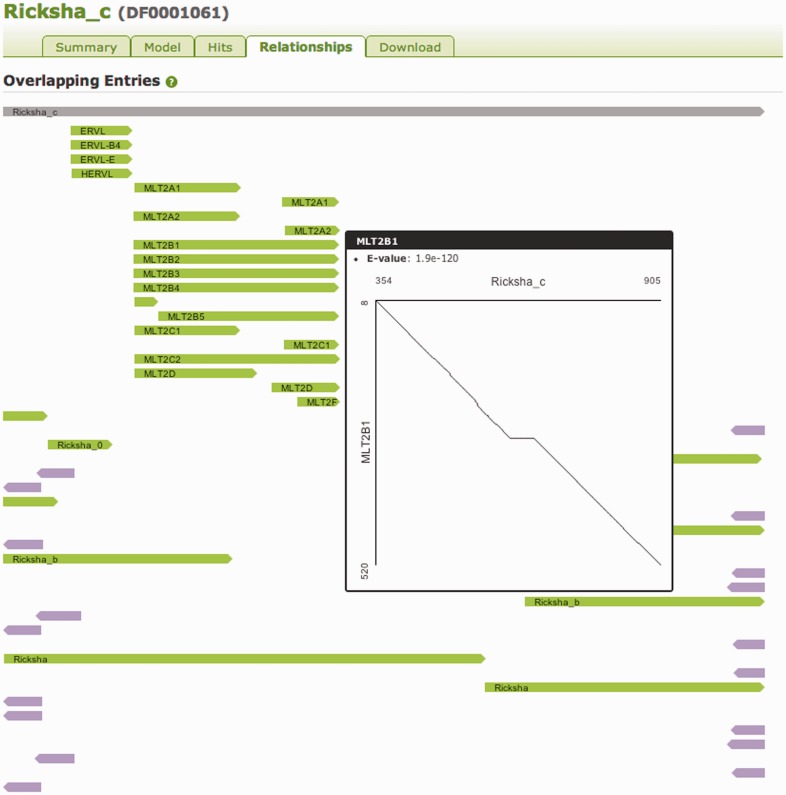
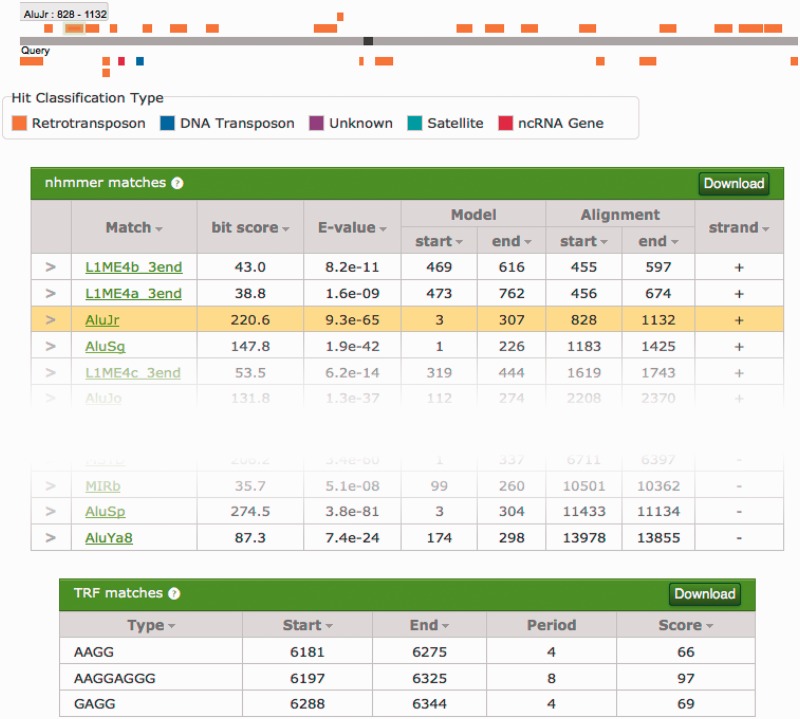
We present a database of repetitive DNA elements, called Dfam (http://dfam.janelia.org). Many genomes contain a large fraction of repetitive DNA, much of which is made up of remnants of transposable elements (TEs). Accurate annotation of TEs enables research into their biology and can shed light on the evolutionary processes that shape genomes. Identification and masking of TEs can also greatly simplify many downstream genome annotation and sequence analysis tasks. The commonly used TE annotation tools RepeatMasker and Censor depend on sequence homology search tools such as cross_match and BLAST variants, as well as Repbase, a collection of known TE families each represented by a single consensus sequence. Dfam contains entries corresponding to all Repbase TE entries for which instances have been found in the human genome. Each Dfam entry is represented by a profile hidden Markov model, built from alignments generated using RepeatMasker and Repbase. When used in conjunction with the hidden Markov model search tool nhmmer, Dfam produces a 2.9% increase in coverage over consensus sequence search methods on a large human benchmark, while maintaining low false discovery rates, and coverage of the full human genome is 54.5%. The website provides a collection of tools and data views to support improved TE curation and annotation efforts. Dfam is also available for download in flat file format or in the form of MySQL table dumps.
Figures
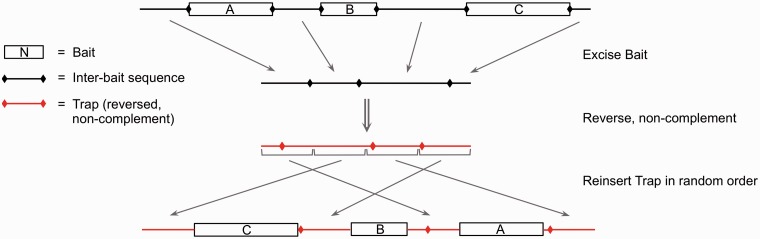
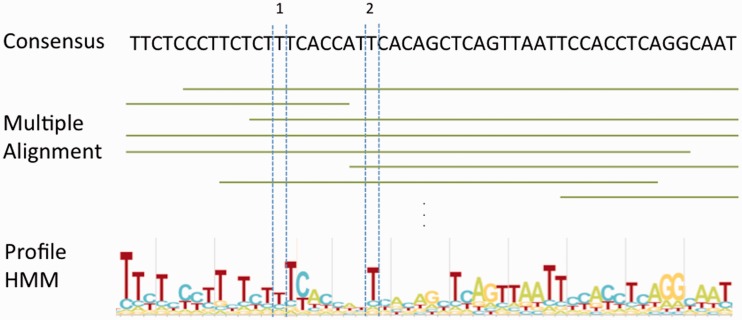
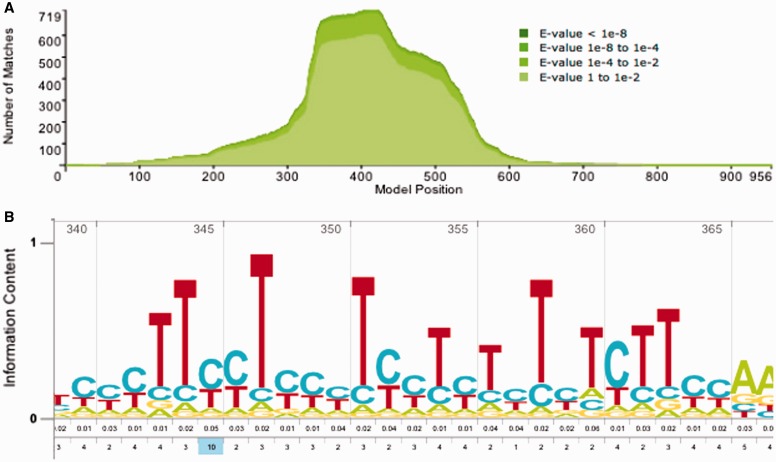
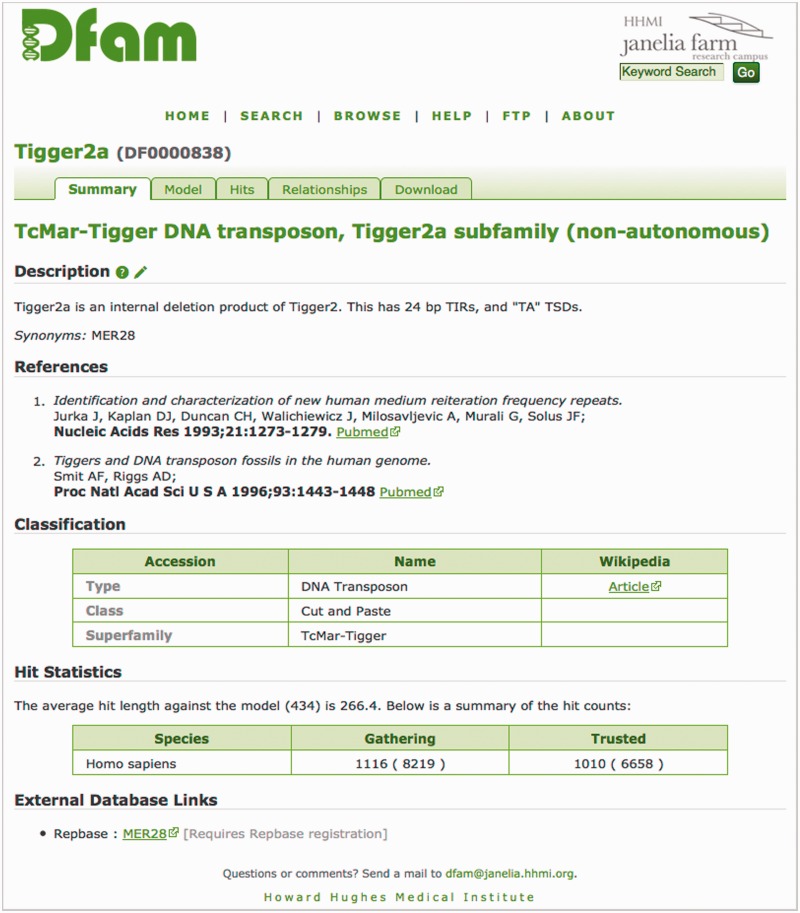
References
-
- Jurka J, Klonowski P, Dagman V, Pelton P. CENSOR—a program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 1996;20:119–121. - PubMed
-
- Smit AFA. 1995. Structure and evolution of mammalian interspersed repeats. Ph.D. Thesis. University of Southern California.
-
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogent. Genome Res. 2005;110:462–467. - PubMed
-
- Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 1998;284:1201–1210. - PubMed
-
- Durbin R, Eddy SR, Krogh A, Mitchison GJ. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
