

iReckon: simultaneous isoform discovery and abundance estimation from RNA-seq data
- PMID: 23204306
- PMCID: PMC3589540
- DOI: 10.1101/gr.142232.112
iReckon: simultaneous isoform discovery and abundance estimation from RNA-seq data
Abstract
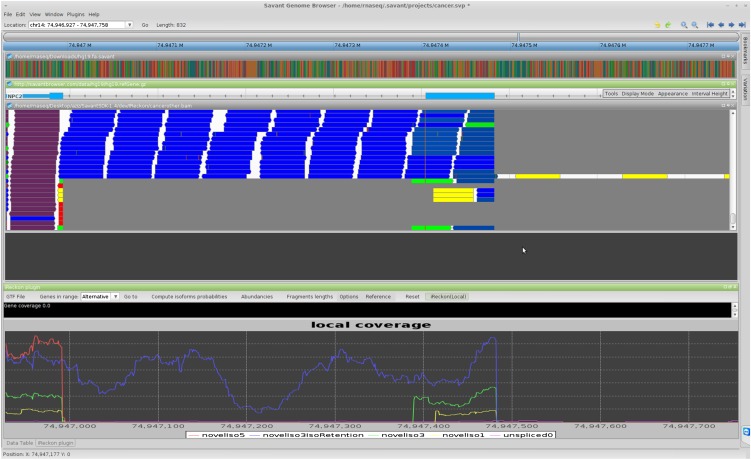
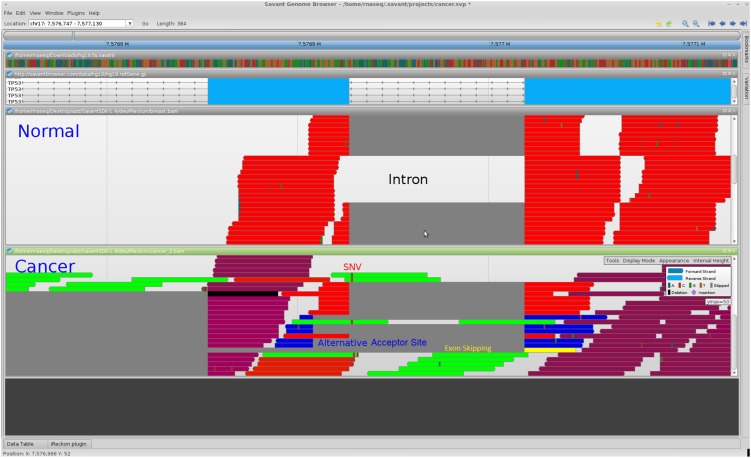
High-throughput RNA sequencing (RNA-seq) promises to revolutionize our understanding of genes and their role in human disease by characterizing the RNA content of tissues and cells. The realization of this promise, however, is conditional on the development of effective computational methods for the identification and quantification of transcripts from incomplete and noisy data. In this article, we introduce iReckon, a method for simultaneous determination of the isoforms and estimation of their abundances. Our probabilistic approach incorporates multiple biological and technical phenomena, including novel isoforms, intron retention, unspliced pre-mRNA, PCR amplification biases, and multimapped reads. iReckon utilizes regularized expectation-maximization to accurately estimate the abundances of known and novel isoforms. Our results on simulated and real data demonstrate a superior ability to discover novel isoforms with a significantly reduced number of false-positive predictions, and our abundance accuracy prediction outmatches that of other state-of-the-art tools. Furthermore, we have applied iReckon to two cancer transcriptome data sets, a triple-negative breast cancer patient sample and the MCF7 breast cancer cell line, and show that iReckon is able to reconstruct the complex splicing changes that were not previously identified. QT-PCR validations of the isoforms detected in the MCF7 cell line confirmed all of iReckon's predictions and also showed strong agreement (r(2) = 0.94) with the predicted abundances.
Figures
References
-
- Berget S 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous